Mechanobiology of Leukocyte Adhesion
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Paxillin Binding to the Cytoplasmic Domain of CD103 Promotes Cell Adhesion and Effector
Author Manuscript Published OnlineFirst on October 11, 2017; DOI: 10.1158/0008-5472.CAN-17-1487 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Paxillin binding to the cytoplasmic domain of CD103 promotes cell adhesion and effector functions for CD8+ resident memory T cells in tumors Ludiane Gauthier1, Stéphanie Corgnac1, Marie Boutet1, Gwendoline Gros1, Pierre Validire2, Georges Bismuth3 and Fathia Mami-Chouaib1 1 INSERM UMR 1186, Integrative Tumor Immunology and Genetic Oncology, Gustave Roussy, EPHE, Fac. de médecine - Univ. Paris-Sud, Université Paris-Saclay, 94805, Villejuif, France 2 Institut Mutualiste Montsouris, Service d’Anatomie pathologique, 75014 Paris, France. 3 INSERM U1016, CNRS UMR8104, Université Paris Descartes, Institut Cochin, 75014 Paris. S Corgnac, M Boutet and G Gros contributed equally to this work. M Boutet current address: Department of Microbiology and Immunology Albert Einstein College of Medecine, NY 10461 USA. Corresponding author: Fathia Mami-Chouaib, INSERM UMR 1186, Gustave Roussy. 39, rue Camille Desmoulins, F-94805 Villejuif. Phone: +33 1 42 11 49 65, Fax: +33 1 42 11 52 88, e-mail: [email protected] and [email protected] Running title: CD103 signaling in human TRM cells Key words: TRM cells, CD103 integrin, T-cell function and signaling, paxillin. Abbreviations: IS: immune synapse; LFA: leukocyte function-associated antigen; FI: fluorescence intensity; mAb: monoclonal antibody; phospho: phosphorylated; Pyk2: proline- rich tyrosine kinase-2; NSCLC: non-small-cell lung carcinoma; r: recombinant; sh-pxn: shorthairpin RNA-paxillin; TCR: T-cell receptor; TIL: tumor-infiltrating lymphocyte; TRM: tissue-resident memory T. -

Human Integrin Alpha L Gene Cdna Clone Plasmid
Human Integrin alpha L Gene cDNA clone plasmid Catalog Number: HG10812-M General Information Plasmid Resuspension protocol Gene : integrin, alpha L (antigen CD11A (p180), lymphocyte function-associated antigen 1; 1.Centrifuge at 5,000×g for 5 min. alpha polypeptide) 2.Carefully open the tube and add 100 l of sterile water to dissolve the DNA. Official Symbol : ITGAL 3.Close the tube and incubate for 10 minutes at room temperature. 4.Briefly vortex the tube and then do a quick spin to concentrate Synonym : CD11A, LFA-1, LFA1A, ITGAL the liquid at the bottom. Speed is less than 5000×g. 5.Store the plasmid at -20 ℃. Source : Human The plasmid is ready for: cDNA Size: 3513bp • Restriction enzyme digestion • PCR amplification RefSeq : NM_002209.2 • E. coli transformation • DNA sequencing Plasmid: pMD-ITGAL E.coli strains for transformation (recommended Description but not limited) Lot : Please refer to the label on the tube Most commercially available competent cells are appropriate for Sequence Description : the plasmid, e.g. TOP10, DH5α and TOP10F´. Identical with the Gene Bank Ref. ID sequence except for the point mutation 2928 A/G not causing the amino acid variation. Vector : pMD18-T Simple Shipping carrier : Each tube contains approximately 10 μg of lyophilized plasmid. Storage : The lyophilized plasmid can be stored at ambient temperature for three months. Quality control : The plasmid is confirmed by full-length sequencing with primers in the sequencing primer list. Sequencing primer list : M13-47 : 5’ GCCAGGGTTTTCCCAGTCACGAC 3’ RV-M : 5’ GAGCGGATAACAATTTCACACAGG 3’ Other M13 primers can also be used as sequencing primers. -

Supplementary Table 1: Adhesion Genes Data Set
Supplementary Table 1: Adhesion genes data set PROBE Entrez Gene ID Celera Gene ID Gene_Symbol Gene_Name 160832 1 hCG201364.3 A1BG alpha-1-B glycoprotein 223658 1 hCG201364.3 A1BG alpha-1-B glycoprotein 212988 102 hCG40040.3 ADAM10 ADAM metallopeptidase domain 10 133411 4185 hCG28232.2 ADAM11 ADAM metallopeptidase domain 11 110695 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 195222 8038 hCG40937.4 ADAM12 ADAM metallopeptidase domain 12 (meltrin alpha) 165344 8751 hCG20021.3 ADAM15 ADAM metallopeptidase domain 15 (metargidin) 189065 6868 null ADAM17 ADAM metallopeptidase domain 17 (tumor necrosis factor, alpha, converting enzyme) 108119 8728 hCG15398.4 ADAM19 ADAM metallopeptidase domain 19 (meltrin beta) 117763 8748 hCG20675.3 ADAM20 ADAM metallopeptidase domain 20 126448 8747 hCG1785634.2 ADAM21 ADAM metallopeptidase domain 21 208981 8747 hCG1785634.2|hCG2042897 ADAM21 ADAM metallopeptidase domain 21 180903 53616 hCG17212.4 ADAM22 ADAM metallopeptidase domain 22 177272 8745 hCG1811623.1 ADAM23 ADAM metallopeptidase domain 23 102384 10863 hCG1818505.1 ADAM28 ADAM metallopeptidase domain 28 119968 11086 hCG1786734.2 ADAM29 ADAM metallopeptidase domain 29 205542 11085 hCG1997196.1 ADAM30 ADAM metallopeptidase domain 30 148417 80332 hCG39255.4 ADAM33 ADAM metallopeptidase domain 33 140492 8756 hCG1789002.2 ADAM7 ADAM metallopeptidase domain 7 122603 101 hCG1816947.1 ADAM8 ADAM metallopeptidase domain 8 183965 8754 hCG1996391 ADAM9 ADAM metallopeptidase domain 9 (meltrin gamma) 129974 27299 hCG15447.3 ADAMDEC1 ADAM-like, -

Impaired Deformability of Erythrocytes and Neutrophils in Children with Newly Diagnosed Insulin-Dependent Diabetes Mellitus
Diabetologia (1999) 42: 865±869 Ó Springer-Verlag 1999 Impaired deformability of erythrocytes and neutrophils in children with newly diagnosed insulin-dependent diabetes mellitus O. Linderkamp1,P.Ruef1, E.P. Zilow1, G.F. Hoffmann2 1 Department of Paediatrics, University of Heidelberg, Germany 2 Department of Paediatrics, University of Marburg, Germany Abstract than in healthy children (3 ± 2%). Deformability of passive neutrophils was greatly decreased in the chil- Aims/hypothesis. Abnormal rheological properties of dren with onset diabetes and moderately reduced in erythrocytes, leucocytes and plasma may have a role the diabetic children who were treated with insulin. in the development of diabetic microangiopathy. We Neutrophil deformation (r = ±0.52) and erythrocyte hypothesized that changed haemorrheological vari- deformation at 0.6 Pa (r = ±0.62) were inversely relat- ables may already be found in children with onset di- ed to haemoglobin A1c. Haematocrit and blood vis- abetes. cosity were increased in the untreated children and Methods. Erythrocyte deformation (rheoscope), neu- in the children treated with insulin for 5 to 8 years. trophil deformation (micropipette), erythrocyte ag- Plasma viscosity and erythrocyte aggregation were gregation, blood and plasma viscosity were measured similar in the three groups of children. in 15 children with insulin-dependent diabetes melli- Conclusion/interpretation. Decreased erythrocyte de- tus before initiation of insulin treatment and 4 to formation at low shear force, increased count of ac- 6 weeks later, 15 diabetic children treated with insulin tive neutrophils and impaired deformability of pas- for 5 to 8 years, 15 healthy children and 15 healthy sive neutrophils may increase the risk for acute cere- adults. -

Human ITGAL Peptide (DAG-P0325) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Human ITGAL peptide (DAG-P0325) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Antigen Description ITGAL encodes the integrin alpha L chain. Integrins are heterodimeric integral membrane proteins composed of an alpha chain and a beta chain. This I-domain containing alpha integrin combines with the beta 2 chain (ITGB2) to form the integrin lymphocyte function-associated antigen-1 (LFA-1), which is expressed on all leukocytes. LFA-1 plays a central role in leukocyte intercellular adhesion through interactions with its ligands, ICAMs 1-3 (intercellular adhesion molecules 1 through 3), and also functions in lymphocyte costimulatory signaling. Two transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jul 2008] Specificity Leukocytes. Nature Synthetic Expression System N/A Purity 70 - 90% by HPLC. Conjugate Unconjugated Sequence Similarities Belongs to the integrin alpha chain family.Contains 7 FG-GAP repeats.Contains 1 VWFA domain. Cellular Localization Membrane. Procedure None Format Liquid Preservative None Storage Shipped at 4°C. Upon delivery aliquot and store at -20°C or -80°C. Avoid repeated freeze / thaw cycles. Information available upon request. ANTIGEN GENE INFORMATION Gene Name ITGAL integrin, alpha L (antigen CD11A (p180), lymphocyte function-associated antigen 1; alpha polypeptide) [ Homo sapiens (human) ] 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative -
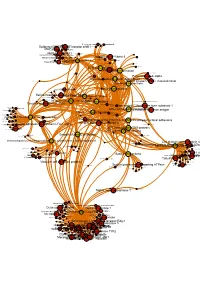
Brcaallwithlabelsintegrina1b1
Receptor-type tyrosine-proteinMuscle- skeletal receptor phosphatase tyrosine protein kinase S Dual specificity mitogen-activated protein kinase kinase 1 Plasminogen activator inhibitor-1 Epidermal growth factor receptor erbB1 Beta amyloid A4 protein MAPDual specificity Epidermalkinase mitogen-activated growth factorCasein proteinreceptor ERK2 kinase and ErbB2kinase; II (HER1 alphaMEK1/2 and (prime) HER2) Casein kinase II alpha Integrin alpha-IIb MAPCasein kinase kinase II beta ERK1 Dual specificity mitogen-activated protein kinase kinase 2 Lysyl oxidase Integrin alpha-IIb/beta-3 Furin Mitogen-activatedSerine/threonine-protein kinase RAF and Dual specificityEpidermal protein mitogen-activated growth proteinkinase; factor kinase kinase receptor 1 (Raf/MEK)ERK1/ERK2 Integrin alpha-V/beta-3 Integrin alpha-V/beta-3 and alpha-IIb/beta 3 Casein kinase II Signal transduction by L1 Integrin alpha-V/beta-6 Casein kinase II alpha/beta Vitronectin receptor alpha Bone morphogenetic protein 2 ECM proteoglycans Bone morphogenetic protein 4 VitronectinElastic fibre formation Peripheral plasma membrane protein CASK Neuropilin-1 Integrin alpha-V/beta-5 Protein kinaseProtein kinase C (PKC) alpha Integrin alpha-2/beta-3 Molecules associated with elastic fibres PKC alpha and beta-2 MER intracellular domain/EGFR extracellular domain chimera Fibronectin receptor alpha Protein kinase C- PKC; classical/novel Syndecan interactions Integrin alpha-2 von Willebrand factorIntegrin alpha-10 Laminin interactions Integrin alpha-11 Fibrinogen beta chain Serine/threonine-proteinVascular -

Effects of Red Blood Cell Aggregation on Microparticle Margination in Human Blood
Effects of Red Blood Cell Aggregation on Microparticle Margination in Human Blood Mark Stroobach Supervised By: Prof. Marianne Fenech Thesis submitted in partial fulfillment of the requirements for the Master of Applied Science in Biomedical Engineering Degree University of Ottawa Ottawa, Ontario, Canada © Mark Stroobach, Ottawa, Canada, 2017 Abstract Margination is the migration of particles in a channel towards the outer walls of the channel. In blood microcirculation, studying the margination of microparticles is important to understand platelet migration and the kinetics of drug delivery. Many new topics in drug delivery research examine the slow release of drugs through micro particles, such as micelles. The margination of such drug carriers is related to tissue absorption and, consequently, to the efficiency of drug delivery. We hypothesized that the intensity of red blood cell (RBC) aggregation will change the level of margination in a cylindrical channel. RBC aggregation is the reversible process of RBCs clumping together over time, under low fluid shear rate. A higher level of aggregation means that this clumping occurs more quickly. The goal of this thesis is to design an experiment that measures the level margination of microparticles and the effect that RBC aggregation has on margination, in a controlled in vitro environment. Fluorescent microparticles were added to human blood preparations. The aggregation properties of the blood preparation were modulated by the addition of a macromolecule (Dextran 500). The blood preparations were injected into PDMS microfluidic devices that were modified to have circular channels in order to better mimic the geometry of physiological microcirculation. We designed a circular microchannel that worked to capture the marginating microparticles and it was found that the level of margination of the microparticles increased with an increase in aggregation of the RBCs. -

ICAM-1 (Phospho Tyr512) Polyclonal Antibody
ICAM-1 Monoclonal Antibody Catalog No : YM1051 Reactivity : Human Applications : WB,IF/ICC Gene Name : ICAM1 Protein Name : Intercellular adhesion molecule 1 Human Gene Id : 3383 Human Swiss Prot P05362 No : Mouse Swiss Prot P13597 No : Immunogen : Purified recombinant human ICAM-1 (N-terminus) protein fragments expressed in E.coli. Specificity : ICAM-1 Monoclonal Antibody detects endogenous levels of ICAM-1 protein. Formulation : Purified mouse monoclonal in buffer containing 0.1M Tris-Glycine (pH 7.4, 150 mM NaCl) with 0.2% sodium azide, 50% glycerol. Source : Mouse Dilution : Western Blot: 1/1000 - 1/2000. Immunofluorescence: 1/100 - 1/500. Not yet tested in other applications. Purification : Affinity purification Concentration : 1 mg/ml Storage Stability : -20°C/1 year Cell Pathway : Cell adhesion molecules (CAMs),Natural killer cell mediated cytotoxicity,Leukocyte transendothelial migration,Viral myocarditis, 1 / 2 Background : intercellular adhesion molecule 1(ICAM1) Homo sapiens This gene encodes a cell surface glycoprotein which is typically expressed on endothelial cells and cells of the immune system. It binds to integrins of type CD11a / CD18, or CD11b / CD18 and is also exploited by Rhinovirus as a receptor. [provided by RefSeq, Jul 2008], Function : function:ICAM proteins are ligands for the leukocyte adhesion protein LFA-1 (integrin alpha-L/beta-2). During leukocyte trans-endothelial migration, ICAM1 engagement promotes the assembly of endothelial apical cups through SGEF and RHOG activation. In case of rhinovirus infection acts as a cellular receptor for the virus.,online information:ICAM-1,online information:Icosahedral capsid structure,online information:Intercellular adhesion molecule entry,polymorphism:Homozygotes with ICAM1-Kalifi Met-56 seem to have an increased risk for cerebral malaria.,PTM:Monoubiquitinated, which is promoted by MARCH9 and leads to endocytosis.,similarity:Belongs to the immunoglobulin superfamily. -

CD11/CD18 (24) FITC FITC Labeled Mouse Monoclonal Antibody
Product Data Sheet IMQ-84601_2015.09-v1 CD11/CD18 (24) FITC FITC Labeled Mouse Monoclonal Antibody Catalog Number: IMQ-84601 Size: 50 µl (1 mg/ml) Class: Monoclonal Type: Antibody Clone: 24 Host / Isotype: Mouse / IgG1 Immunogen: Fibronectin-purified human monocytes Myeloma / fusion Cells from immunized Balb/c mice were fused with the Sp2/0-Ag.14 myeloma partners: cell line Species Reactivity: Human Specificity: This antibody recognises integrin alpha L,M,X/Beta 2 (CD11/CD18). Integrins are heterodimeric cell surface receptors composed of alpha and beta subunits which mediate cell-cell and cell-extracellular matrix attachments. Integrin beta2 (CD18) associates with integrin alpha L (CD11a) to form the leukocyte function- associated antigen-1 (LFA-1) with integrin alpha M (CD11b) to form Complement Receptor 3 (CR3) and with integrin alpha X (CD11c) to form Complement Receptor 4 (CR4). Each integrin receptor can bind various Intercellular adhesion molecules (ICAMs). Aberrant integrin expression has been found in many epithelial tumours. Changes in integrin expression have been shown to be important for the growth and early metastatic capacity of melanoma cells. Purification: Purified on protein A from tissue culture supernatant. Format: Purified IgG conjugated to Fluoroscein using Innova Biosciences Lightning- Link®, supplied in Phosphate buffered saline (PBS) containing 0.09% Sodium azide Applications: Flow Cytometry, Immunocytochemistry, Immunofluorescence Dilutions: Optimal antibody dilution should be determined by titration Storage: Store stock solution of the antibody at 2-8 °C in the dark. Do not freeze. References: Hogg N, et al. An anti-human monocyte/macrophage monoclonal antibody, reacting most strongly with macrophages in lymphoid tissue. -

PDF Download
Integrin β2 Polyclonal Antibody Catalog No : YT2369 Reactivity : Human,Mouse,Rat Applications : IF/ICC,ELISA Gene Name : ITGB2 Protein Name : Integrin beta-2 Human Gene Id : 3689 Human Swiss Prot P05107 No : Mouse Gene Id : 16414 Mouse Swiss Prot P11835 No : Immunogen : The antiserum was produced against synthesized peptide derived from human CD18/ITGB2. AA range:720-769 Specificity : Integrin β2 Polyclonal Antibody detects endogenous levels of Integrin β2 protein. Formulation : Liquid in PBS containing 50% glycerol, 0.5% BSA and 0.02% sodium azide. Source : Rabbit Dilution : Immunofluorescence: 1/200 - 1/1000. ELISA: 1/5000. Not yet tested in other applications. Purification : The antibody was affinity-purified from rabbit antiserum by affinity- chromatography using epitope-specific immunogen. Concentration : 1 mg/ml Storage Stability : -20°C/1 year Molecularweight : 84782 1 / 2 Cell Pathway : Cell adhesion molecules (CAMs),Natural killer cell mediated cytotoxicity,Leukocyte transendothelial migration,Regulates Actin and Cytoskeleton,Viral myocarditis, Background : integrin subunit beta 2(ITGB2) Homo sapiens This gene encodes an integrin beta chain, which combines with multiple different alpha chains to form different integrin heterodimers. Integrins are integral cell-surface proteins that participate in cell adhesion as well as cell-surface mediated signalling. The encoded protein plays an important role in immune response and defects in this gene cause leukocyte adhesion deficiency. Alternative splicing results in multiple transcript variants. [provided by RefSeq, Dec 2014], Function : disease:Defects in ITGB2 are the cause of leukocyte adhesion deficiency type I (LAD1) [MIM:116920]. LAD1 patients have recurrent bacterial infections and their leukocytes are deficient in a wide range of adhesion-dependent functions.,function:Integrin alpha-L/beta-2 is a receptor for ICAM1, ICAM2, ICAM3 and ICAM4. -

Mean Red Cell and Platelet Volume and Blood Cells Aggregation in Children with Inflammatory Bowel Diseases
Research Article Clinics in Surgery Published: 19 Jul, 2018 Mean Red Cell and Platelet Volume and Blood Cells Aggregation in Children with Inflammatory Bowel Diseases Grigory Ya Levin*, Alexandra N Popovicheva, Larisa N Sosnina, El’vira N Fedulova and Yury A Sheremet’ev Department of Gravitation Surgery and Hemodialysis, Federal State Budgetary Institution of the Ministry of Health of Russian Federation, Russia Abstract Background: It has been discussed in literature whether platelet and erythrocyte indexes can serve as biomarkers of activity of Inflammatory Bowel Diseases (IBD). However, how these indexes influence functional properties of platelets and erythrocytes, primarily their aggregation capacity in children with IBD, remains unclear. Objectives: The objective of the present research was to study the association between the changes in platelet and erythrocyte indexes (MPV, PDW, MCV, and RDW) blood cells aggregation during the course of therapy of children with IBD. Methods: The study included 50 patients of both sex ages 6 to 17, 25 patients with UC, and 25 patients with CD. The diagnosis was based on a complex examination including endoscopic examination of the intestinal mucosa with a morphological analysis of biopsies. Spontaneous (shear-induced) aggregation of platelets and erythrocytes was studied using a rheoscope designed according to the method. Results: It was shown that the mean platelet volume and the Platelet Distribution Width (PDW) significantly decrease at IBD, whilst Erythrocyte Distribution Width (RDW) and mean erythrocyte OPEN ACCESS volume increase. A strong correlation between RDW and IBD severity as well as a negative correlation *Correspondence: between MPV and IBD severity were revealed. For the first time it has been established that, with Levin Grigory Yakovlevich, a reduced volume, platelets and erythrocytes retain their functional properties, in particular their Department of Gravitation Surgery and aggregation activity. -

Physical Charaterization of Red Blood Cell Aggregation Daniel Amadeus Dominic Flormann
Physical charaterization of red blood cell aggregation Daniel Amadeus Dominic Flormann To cite this version: Daniel Amadeus Dominic Flormann. Physical charaterization of red blood cell aggregation. Biological Physics [physics.bio-ph]. Universität des Saarlandes, 2017. English. NNT : 2017GREAY002. tel- 01577838 HAL Id: tel-01577838 https://tel.archives-ouvertes.fr/tel-01577838 Submitted on 28 Aug 2017 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. THÈSE Pour obtenir le grade de DOCTEUR DE LA COMMUNAUTÉ UNIVERSITÉ GRENOBLE ALPES préparée dans le cadre d’une cotutelle entre la Communauté Université Grenoble Alpes et l’Universität des Saarlandes Spécialité : Physique pour les sciences du vivant Arrêté ministériel : 25 mai 2016 Présentée par Daniel Amadeus Dominic Flormann Thèse dirigée par M. Thomas Podgorski et M. Christian Wagner préparée au sein du Laboratoire Interdisciplinaire de Physique, Grenoble et de Experimentalphysik, Saarbrücken dans les Écoles doctorales de Physique de l’Université Grenoble Alpes et de Dekanat der Universität des Saarlandes