Compound Heterozygous Alterations in Intraflagellar Transport Protein CLUAP1 in a Child with A
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Educational Paper Ciliopathies
Eur J Pediatr (2012) 171:1285–1300 DOI 10.1007/s00431-011-1553-z REVIEW Educational paper Ciliopathies Carsten Bergmann Received: 11 June 2011 /Accepted: 3 August 2011 /Published online: 7 September 2011 # The Author(s) 2011. This article is published with open access at Springerlink.com Abstract Cilia are antenna-like organelles found on the (NPHP) . Ivemark syndrome . Meckel syndrome (MKS) . surface of most cells. They transduce molecular signals Joubert syndrome (JBTS) . Bardet–Biedl syndrome (BBS) . and facilitate interactions between cells and their Alstrom syndrome . Short-rib polydactyly syndromes . environment. Ciliary dysfunction has been shown to Jeune syndrome (ATD) . Ellis-van Crefeld syndrome (EVC) . underlie a broad range of overlapping, clinically and Sensenbrenner syndrome . Primary ciliary dyskinesia genetically heterogeneous phenotypes, collectively (Kartagener syndrome) . von Hippel-Lindau (VHL) . termed ciliopathies. Literally, all organs can be affected. Tuberous sclerosis (TSC) . Oligogenic inheritance . Modifier. Frequent cilia-related manifestations are (poly)cystic Mutational load kidney disease, retinal degeneration, situs inversus, cardiac defects, polydactyly, other skeletal abnormalities, and defects of the central and peripheral nervous Introduction system, occurring either isolated or as part of syn- dromes. Characterization of ciliopathies and the decisive Defective cellular organelles such as mitochondria, perox- role of primary cilia in signal transduction and cell isomes, and lysosomes are well-known -

Ciliopathiesneuromuscularciliopathies Disorders Disorders Ciliopathiesciliopathies
NeuromuscularCiliopathiesNeuromuscularCiliopathies Disorders Disorders CiliopathiesCiliopathies AboutAbout EGL EGL Genet Geneticsics EGLEGL Genetics Genetics specializes specializes in ingenetic genetic diagnostic diagnostic testing, testing, with with ne nearlyarly 50 50 years years of of clinical clinical experience experience and and board-certified board-certified labor laboratoryatory directorsdirectors and and genetic genetic counselors counselors reporting reporting out out cases. cases. EGL EGL Genet Geneticsics offers offers a combineda combined 1000 1000 molecular molecular genetics, genetics, biochemical biochemical genetics,genetics, and and cytogenetics cytogenetics tests tests under under one one roof roof and and custom custom test testinging for for all all medically medically relevant relevant genes, genes, for for domestic domestic andand international international clients. clients. EquallyEqually important important to to improving improving patient patient care care through through quality quality genetic genetic testing testing is is the the contribution contribution EGL EGL Genetics Genetics makes makes back back to to thethe scientific scientific and and medical medical communities. communities. EGL EGL Genetics Genetics is is one one of of only only a afew few clinical clinical diagnostic diagnostic laboratories laboratories to to openly openly share share data data withwith the the NCBI NCBI freely freely available available public public database database ClinVar ClinVar (>35,000 (>35,000 variants variants on on >1700 >1700 genes) genes) and and is isalso also the the only only laboratory laboratory with with a a frefree oen olinnlein dea dtabtaabsaes (eE m(EVmCVlaCslas)s,s f)e, afetuatruinrgin ag vaa vraiarniatn ctl acslasisfiscifiactiaotino sne saercahrc ahn adn rde rpeoprot rrte rqeuqeuset sint tinetrefarcfaec, ew, hwichhic fha cfailcitialiteatse rsa praidp id interactiveinteractive curation curation and and reporting reporting of of variants. -
Shimada 2017.Pdf
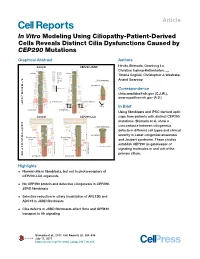
Article In Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations Graphical Abstract Authors Control CEP290-JSRD Hiroko Shimada, Quanlong Lu, Christine Insinna-Kettenhofen, ..., Cilium GPR161 Cilium ADCY3 Tiziana Cogliati, Christopher J. Westlake, Smo ARL13B F CEP290 Cell membrane Anand Swaroop I Gli2 B Cytoplasm R Other proteins O Correspondence Axoneme B Axoneme [email protected] (C.J.W.), L A [email protected] (A.S.) S Cell membrane Cell membrane T Vesicles In Brief Cytoplasm Cytoplasm Using fibroblasts and iPSC-derived optic Control CEP290-LCA cups from patients with distinct CEP290 mutations, Shimada et al. show a P H Cell membrane concordance between ciliogenesis O T ? defects in different cell types and clinical Axoneme ? O ? Axoneme ? Cytoplasm severity in Leber congenital amaurosis R ? Connecting Connecting ? E Cilium Cilium ? and Joubert syndrome. These studies Cell membrane C Cell membrane E establish CEP290 as gatekeeper of P Docked mother centriole T ?? ? ? signaling molecules in and out of the O primary cilium. R Cytoplasm Cytoplasm Highlights d Normal cilia in fibroblasts, but not in photoreceptors of CEP290-LCA organoids d No CEP290 protein and defective ciliogenesis in CEP290- JSRD fibroblasts d Selective reduction in ciliary localization of ARL13B and ADCY3 in JSRD fibroblasts d Cilia defects in JSRD fibroblasts affect Smo and GPR161 transport in Hh signaling Shimada et al., 2017, Cell Reports 20, 384–396 July 11, 2017 http://dx.doi.org/10.1016/j.celrep.2017.06.045 Cell Reports Article In Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations Hiroko Shimada,1 Quanlong Lu,2 Christine Insinna-Kettenhofen,2 Kunio Nagashima,3 Milton A. -

Unraveling the Genetics of Joubert and Meckel-Gruber Syndromes
Journal of Pediatric Genetics 3 (2014) 65–78 65 DOI 10.3233/PGE-14090 IOS Press Unraveling the genetics of Joubert and Meckel-Gruber syndromes Katarzyna Szymanska, Verity L. Hartill and Colin A. Johnson∗ Department of Ophthalmology and Neuroscience, University of Leeds, Leeds, UK Received 27 May 2014 Revised 11 July 2014 Accepted 14 July 2014 Abstract. Joubert syndrome (JBTS) and Meckel-Gruber syndrome (MKS) are recessive neurodevelopmental conditions caused by mutations in proteins that are structural or functional components of the primary cilium. In this review, we provide an overview of their clinical diagnosis, management and molecular genetics. Both have variable phenotypes, extreme genetic heterogeneity, and display allelism both with each other and other ciliopathies. Recent advances in genetic technology have significantly improved diagnosis and clinical management of ciliopathy patients, with the delineation of some general genotype-phenotype correlations. We highlight those that are most relevant for clinical practice, including the correlation between TMEM67 mutations and the JBTS variant phenotype of COACH syndrome. The subcellular localization of the known MKS and JBTS proteins is now well-described, and we discuss some of the contemporary ideas about ciliopathy disease pathogenesis. Most JBTS and MKS proteins localize to a discrete ciliary compartment called the transition zone, and act as structural components of the so-called “ciliary gate” to regulate the ciliary trafficking of cargo proteins or lipids. Cargo proteins include enzymes and transmembrane proteins that mediate intracellular signaling. The disruption of transition zone function may contribute to the ciliopathy phenotype by altering the composition of the ciliary membrane or axoneme, with impacts on essential developmental signaling including the Wnt and Shh pathways as well as the regulation of secondary messengers such as inositol-1,4,5-trisphosphate (InsP3) and cyclic adenosine monophosphate (cAMP). -

Prenatal Exome Sequencing in Anomalous Fetuses: New Opportunities and Challenges
© American College of Medical Genetics and Genomics ORIGINAL RESEARCH ARTICLE Prenatal exome sequencing in anomalous fetuses: new opportunities and challenges Neeta L. Vora, MD1, Bradford Powell, MD, PhD2, Alicia Brandt, MS2, Natasha Strande, PhD3,4, Emily Hardisty, MS, CGC1, Kelly Gilmore, MS, CGC1, Ann Katherine M. Foreman, MS, CGC2,5, Kirk Wilhelmsen, MD, PhD6, Chris Bizon, PhD6, Jason Reilly, BA6, Phil Owen, BS6, Cynthia M. Powell, MD, MS2,7, Debra Skinner, PhD, MA8, Christine Rini, PhD9, Anne D. Lyerly, MD, MA10, Kim A. Boggess, MD1, Karen Weck, MD3,4, Jonathan S. Berg, MD, PhD2 and James P. Evans, MD, PhD2,10 Purpose: We investigated the diagnostic and clinical performance the following genes: COL1A1, MUSK, KCTD1, RTTN, TMEM67, of exome sequencing in fetuses with sonographic abnormalities PIEZO1 and DYNC2H1. One additional case revealed a de novo with normal karyotype and microarray and, in some cases, normal nonsense mutation in a novel candidate gene (MAP4K4). gene-specific sequencing. The perceived likelihood that exome sequencing would explain Methods: Exome sequencing was performed on DNA from 15 the results (5.2 on a 10-point scale) was higher than the anomalous fetuses and from the peripheral blood of their approximately 30% diagnostic yield discussed in pretest counseling. parents. Parents provided consent to be informed of diagnostic Conclusion: Exome sequencing had diagnostic utility in a highly results in the fetus, medically actionable findings in the parents, select population of fetuses where a genetic diagnosis was highly and their identification as carrier couples for significant auto- suspected. Challenges related to genetics literacy and variant somal recessive conditions. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

SDCCAG8 Interacts with RAB Effector Proteins RABEP2 and ERC1 and Is Required for Hedgehog Signaling
SDCCAG8 Interacts with RAB Effector Proteins RABEP2 and ERC1 and Is Required for Hedgehog Signaling Airik, Rannar; Schueler, Markus; Airik, Merlin; Cho, Jang; Ulanowicz, Kelsey A; Porath, Jonathan D; Hurd, Toby W; Bekker-Jensen, Simon; Schrøder, Jacob Morville; Andersen, Jens S.; Hildebrandt, Friedhelm Published in: P L o S One DOI: 10.1371/journal.pone.0156081 Publication date: 2016 Document version Publisher's PDF, also known as Version of record Document license: CC BY Citation for published version (APA): Airik, R., Schueler, M., Airik, M., Cho, J., Ulanowicz, K. A., Porath, J. D., Hurd, T. W., Bekker-Jensen, S., Schrøder, J. M., Andersen, J. S., & Hildebrandt, F. (2016). SDCCAG8 Interacts with RAB Effector Proteins RABEP2 and ERC1 and Is Required for Hedgehog Signaling. P L o S One, 11(5), [e0156081]. https://doi.org/10.1371/journal.pone.0156081 Download date: 04. Oct. 2021 RESEARCH ARTICLE SDCCAG8 Interacts with RAB Effector Proteins RABEP2 and ERC1 and Is Required for Hedgehog Signaling Rannar Airik1¤‡*, Markus Schueler1, Merlin Airik1, Jang Cho1, Kelsey A. Ulanowicz2, Jonathan D. Porath1, Toby W. Hurd3, Simon Bekker-Jensen4, Jacob M. Schrøder5, Jens S. Andersen5, Friedhelm Hildebrandt1,6‡* 1 Department of Medicine, Division of Nephrology, Boston Children’s Hospital, Boston, Massachusetts, United States of America, 2 Department of Pediatrics, Division of Nephrology, Children’s Hospital of a11111 Pittsburgh of UPMC, Pittsburgh, Pennsylvania, United States of America, 3 Medical Research Council Human Genetics Unit, Institute of -

Ciliopathy-Associated Gene Cc2d2a Promotes Assembly of Subdistal Appendages on the Mother Centriole During Cilia Biogenesis
ARTICLE Received 7 Apr 2014 | Accepted 23 May 2014 | Published 20 Jun 2014 DOI: 10.1038/ncomms5207 Ciliopathy-associated gene Cc2d2a promotes assembly of subdistal appendages on the mother centriole during cilia biogenesis Shobi Veleri1, Souparnika H. Manjunath1, Robert N. Fariss2, Helen May-Simera1, Matthew Brooks1, Trevor A. Foskett1, Chun Gao2, Teresa A. Longo1, Pinghu Liu3, Kunio Nagashima4, Rivka A. Rachel1, Tiansen Li1, Lijin Dong3 & Anand Swaroop1 The primary cilium originates from the mother centriole and participates in critical functions during organogenesis. Defects in cilia biogenesis or function lead to pleiotropic phenotypes. Mutations in centrosome-cilia gene CC2D2A result in Meckel and Joubert syndromes. Here we generate a Cc2d2a À / À mouse that recapitulates features of Meckel syndrome including embryonic lethality and multiorgan defects. Cilia are absent in Cc2d2a À / À embryonic node and other somatic tissues; disruption of cilia-dependent Shh signalling appears to underlie exencephaly in mutant embryos. The Cc2d2a À / À mouse embryonic fibroblasts (MEFs) lack cilia, although mother centrioles and pericentriolar pro- teins are detected. Odf2, associated with subdistal appendages, is absent and ninein is reduced in mutant MEFs. In Cc2d2a À / À MEFs, subdistal appendages are lacking or abnormal by transmission electron microscopy. Consistent with this, CC2D2A localizes to subdistal appendages by immuno-EM in wild-type cells. We conclude that CC2D2A is essential for the assembly of subdistal appendages, which anchor cytoplasmic microtubules and prime the mother centriole for axoneme biogenesis. 1 Neurobiology-Neurodegeneration and Repair Laboratory, National Eye Institute, NIH, Bethesda, Maryland 20892, USA. 2 Biological Imaging Core, National Eye Institute, NIH, Bethesda, Maryland 20892, USA. -

Ciliopathies Gene Panel
Ciliopathies Gene Panel Contact details Introduction Regional Genetics Service The ciliopathies are a heterogeneous group of conditions with considerable phenotypic overlap. Levels 4-6, Barclay House These inherited diseases are caused by defects in cilia; hair-like projections present on most 37 Queen Square cells, with roles in key human developmental processes via their motility and signalling functions. Ciliopathies are often lethal and multiple organ systems are affected. Ciliopathies are London, WC1N 3BH united in being genetically heterogeneous conditions and the different subtypes can share T +44 (0) 20 7762 6888 many clinical features, predominantly cystic kidney disease, but also retinal, respiratory, F +44 (0) 20 7813 8578 skeletal, hepatic and neurological defects in addition to metabolic defects, laterality defects and polydactyly. Their clinical variability can make ciliopathies hard to recognise, reflecting the ubiquity of cilia. Gene panels currently offer the best solution to tackling analysis of genetically Samples required heterogeneous conditions such as the ciliopathies. Ciliopathies affect approximately 1:2,000 5ml venous blood in plastic EDTA births. bottles (>1ml from neonates) Ciliopathies are generally inherited in an autosomal recessive manner, with some autosomal Prenatal testing must be arranged dominant and X-linked exceptions. in advance, through a Clinical Genetics department if possible. Referrals Amniotic fluid or CV samples Patients presenting with a ciliopathy; due to the phenotypic variability this could be a diverse set should be sent to Cytogenetics for of features. For guidance contact the laboratory or Dr Hannah Mitchison dissecting and culturing, with ([email protected]) / Prof Phil Beales ([email protected]) instructions to forward the sample to the Regional Molecular Genetics Referrals will be accepted from clinical geneticists and consultants in nephrology, metabolic, laboratory for analysis respiratory and retinal diseases. -

Phosphoinositide 3-Kinase-C2α Regulates Polycystin-2 Ciliary Entry
BASIC RESEARCH www.jasn.org Phosphoinositide 3-Kinase-C2a Regulates Polycystin-2 Ciliary Entry and Protects against Kidney Cyst Formation † Irene Franco,* Jean Piero Margaria,* Maria Chiara De Santis,* Andrea Ranghino, ‡ Daniel Monteyne, Marco Chiaravalli,§ Monika Pema,§ Carlo Cosimo Campa,* ‡| Edoardo Ratto,* Federico Gulluni,* David Perez-Morga, Stefan Somlo,¶ Giorgio R. Merlo,* Alessandra Boletta,§ and Emilio Hirsch* *Molecular Biotechnology Center, Department of Molecular Biotechnology and Health Sciences, University of Torino, Turin, Italy; †Renal Transplantation Center “A. Vercellone”, Division of Nephrology, Dialysis and Transplantation, Department of Medical Sciences, Città della Salute e della Scienza, Hospital and Research Center for Experimental Medicine (CeRMS) and Center for Molecular Biotechnology, University of Torino, Turin, Italy; ‡Laboratoire de Parasitologie Moléculaire, Institut de Biologie et de Médecine Moléculaires (IBMM), Université Libre de Bruxelles, Gosselies, Charleroi, Belgium; §Division of Genetics and Cell Biology, Dibit San Raffaele Scientific Institute, Milan, Italy; |Center for Microscopy and Molecular Imaging (CMMI), Université Libre de Bruxelles, Gosselies, Belgium; and ¶Section of Nephrology, Yale University School of Medicine, New Haven, Connecticut. ABSTRACT Signaling from the primary cilium regulates kidney tubule development and cyst formation. However, the mechanism controlling targeting of ciliary components necessary for cilium morphogenesis and signaling is largely unknown. Here, we studied the function of class II phosphoinositide 3-kinase-C2a (PI3K-C2a)inrenal tubule-derived inner medullary collecting duct 3 cells and show that PI3K-C2a resides at the recycling endo- some compartment in proximity to the primary cilium base. In this subcellular location, PI3K-C2a controlled the activation of Rab8, a key mediator of cargo protein targeting to the primary cilium. -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig.