I* Carrike 7Rse Separation 07 Cobalt Ii
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Stable Isotopes of Cobalt Properties of Cobalt
Stable Isotopes of Cobalt Isotope Z(p) N(n) Atomic Mass Natural Abundance Nuclear Spin Co-59 27 32 58.93320 100.00% 7/2- Cobalt was discovered in 1735 by Georg Brandt. Its name derives from the German word kobald, meaning "goblin" or "evil spirit." Minerals containing cobalt were used by the early civilizations of Egypt and Mesopotamia for coloring glass deep blue. Cobalt oxide is used today to add a pink or blue color to glass. It is also an important trace element in soils and necessary for animal nutrition. The most important modern use of cobalt is in the manufacture of various wear-resistant and superalloys. Its alloys have shown high resistance to corrosion and oxidation at high temperatures. Radioactive Cobalt-60 is used in radiography and in the sterilization of food. A silvery-white, shining, hard, ductile, somewhat malleable metal, cobalt is also ferromagnetic, with permeability two-thirds that of iron. It has exceptional magnetic properties in alloys. It is attached by dilute hydrochloric and sulfuric acids. It corrodes readily in air, and it has unusual coordinating properties, especially the trivalent ion. It is noncombustible except in powder form. Cobalt occurs in two allotropic modifications over a wide range of temperatures: the crystalline close-packed- hexagonal form is known as the alpha form, which turns into the beta (or gamma) form above 417 ºC. In finely powdered form, cobalt ignites spontaneously in air. Reactions with acetylene and bromine pentafluoride proceed to incandescence and can become violent. The metal is moderately toxic by ingestion. Inhalation of dusts can damage lungs. -

Annual Report 1951 National Bureau of Standards
Annual Report 1951 National Bureau of Standards Miscellaneous Publication 204 UNITED STATES DEPARTMENT OF COMMERCE Charles Sawyer, Secretary NATIONAL BUREAU OF STANDARDS A. V. Astint, Director Annual Report 1951 National Bureau of Standards For sale by the Superintendent of Documents, U. S. Government Printing Office Washington 2 5, D. C. Price 50 cents CONTENTS Page 1. General Review 1 2. Electricity 16 Beam intensification in a high-voltage oscillograph 17, Low-temperature dry cells 17, High-rate batteries 17, Battery additives 18. 3. Optics and Metrology 18 The kinorama 19, Measurement of visibility for aircraft 20, Antisubmarine aircraft searchlights 20, Resolving power chart 20, Refractivity 21, Thermal expansivity of aluminum alloys 21. 4. Heat and Power . 21 Thermodynamic properties of materials 22, Synthetic rubber and other high polymers 23, Combustors for jet engines 24, Temperature and composition of flames 25, Engine "knock" 25, Low-temperature physics 26, Medical physics instrumentation 28. 5. Atomic and Radiation Physics 28 Atomic standard of length 29, Magnetic moment of the proton 29, Spectra of artificial elements 31, Photoconductivity of semiconductors 31, Radiation detecting instruments 32, Protection against radiation 32, X-ray equipment 33, Atomic and molecular ions 35, Electron physics 35, Tables of nuclear data 35, Atomic energy levels 36. 6. Chemistry 36 Radioactive carbohydrates 36, Dextran as a substitute for blood plasma 37, Acidity and basicity in organic solvents 37, Interchangeability of fuel gases 38, Los Angeles "smog" 39, Infrared spectra of alcohols 39, Electrodeposi- tion 39, Development of analytical methods 40, Physical constants 42. 7. Mechanics 42 Turbulent flow 43, Turbulence at supersonic speeds 43, Dynamic properties of materials 43, High-frequency vibrations 44, Hearing loss 44, Physical properties by sonic methods 44, Water waves 46, Density currents 46, Precision weighing 46, Viscosity of gases 46, Evaporated thin films 47. -

Chapter 2 Atoms, Molecules and Ions
Chapter 2 Atoms, Molecules and Ions PRACTICING SKILLS Atoms:Their Composition and Structure 1. Fundamental Particles Protons Electrons Neutrons Electrical Charges +1 -1 0 Present in nucleus Yes No Yes Least Massive 1.007 u 0.00055 u 1.007 u 3. Isotopic symbol for: 27 (a) Mg (at. no. 12) with 15 neutrons : 27 12 Mg 48 (b) Ti (at. no. 22) with 26 neutrons : 48 22 Ti 62 (c) Zn (at. no. 30) with 32 neutrons : 62 30 Zn The mass number represents the SUM of the protons + neutrons in the nucleus of an atom. The atomic number represents the # of protons, so (atomic no. + # neutrons)=mass number 5. substance protons neutrons electrons (a) magnesium-24 12 12 12 (b) tin-119 50 69 50 (c) thorium-232 90 142 90 (d) carbon-13 6 7 6 (e) copper-63 29 34 29 (f) bismuth-205 83 122 83 Note that the number of protons and electrons are equal for any neutral atom. The number of protons is always equal to the atomic number. The mass number equals the sum of the numbers of protons and neutrons. Isotopes 7. Isotopes of cobalt (atomic number 27) with 30, 31, and 33 neutrons: 57 58 60 would have symbols of 27 Co , 27 Co , and 27 Co respectively. Chapter 2 Atoms, Molecules and Ions Isotope Abundance and Atomic Mass 9. Thallium has two stable isotopes 203 Tl and 205 Tl. The more abundant isotope is:___?___ The atomic weight of thallium is 204.4 u. The fact that this weight is closer to 205 than 203 indicates that the 205 isotope is the more abundant isotope. -

Iron Isotope Cosmochemistry Kun Wang Washington University in St
Washington University in St. Louis Washington University Open Scholarship All Theses and Dissertations (ETDs) 12-2-2013 Iron Isotope Cosmochemistry Kun Wang Washington University in St. Louis Follow this and additional works at: https://openscholarship.wustl.edu/etd Part of the Earth Sciences Commons Recommended Citation Wang, Kun, "Iron Isotope Cosmochemistry" (2013). All Theses and Dissertations (ETDs). 1189. https://openscholarship.wustl.edu/etd/1189 This Dissertation is brought to you for free and open access by Washington University Open Scholarship. It has been accepted for inclusion in All Theses and Dissertations (ETDs) by an authorized administrator of Washington University Open Scholarship. For more information, please contact [email protected]. WASHINGTON UNIVERSITY IN ST. LOUIS Department of Earth and Planetary Sciences Dissertation Examination Committee: Frédéric Moynier, Chair Thomas J. Bernatowicz Robert E. Criss Bruce Fegley, Jr. Christine Floss Bradley L. Jolliff Iron Isotope Cosmochemistry by Kun Wang A dissertation presented to the Graduate School of Arts and Sciences of Washington University in partial fulfillment of the requirements for the degree of Doctor of Philosophy December 2013 St. Louis, Missouri Copyright © 2013, Kun Wang All rights reserved. Table of Contents LIST OF FIGURES ....................................................................................................................... vi LIST OF TABLES ........................................................................................................................ -

Załącznik Nr 3 Do Wniosku O Przeprowadzenie Postępowania Habilitacyjnego
Załącznik nr 3 do wniosku o przeprowadzenie postępowania habilitacyjnego Author's review of his own writings 1. Jan Kurpeta 2. Posiadane dyplomy, stopnie naukowe Degree of Doctor of Philosophy in physics, University of Warsaw, Faculty of Physics, year 1999, title of dissertation: „Properties of the neutron-rich nuclei at the edge of known nuclei area.” Master of Science and Engineering, Warsaw University of Technology, Faculty of Technical Physics and Applied Mathematics, year 1993, title of master's thesis: „Influence of nucleon effective mass on single particle motion in the deformed nuclei.” 3. Informacje o dotychczasowym zatrudnieniu w jednostkach naukowych University of Warsaw, Faculty of Physics, Institute of Experimental Physics, Nuclear Spectroscopy Division, assistant professor since15 February 2001 University of Leuven (Belgium), Instituut voor Kern- en Stralingsfysica, post-doc position from 1 September 1998 to 31 October 1999 University of Jyväskylä (Finland), Faculty of Physics, scholarship from Center for International Mobility, PhD studies from 3 February to 15 December 1995 University of Warsaw, Faculty of Physics, Institute of Experimental Physics, Nuclear Spectroscopy Division, PhD studies from October 1993 to September 1998 Space Research Center Polish Academy of Sciences in Warsaw, Department of Planetary Geodesy, part time job for 6 months in 1993 1 4. Wskazanie osiągnięcia* wynikającego z art. 16 ust. 2 ustawy z dnia 14 marca 2003 r. o stopniach naukowych i tytule naukowym oraz o stopniach i tytule w zakresie sztuki (Dz. U. nr 65, poz. 595 ze zm.): a) tytuł osiągnięcia naukowego Structure of exotic, neutron-rich fission fragments of mass around A = 110. b) Jednotematyczny cykl publikacji przedstawiających osiągnięcie naukowe [A1] J. -

74 Dauphas Et Al EPSL 2014.Pdf
Earth and Planetary Science Letters 398 (2014) 127–140 Contents lists available at ScienceDirect Earth and Planetary Science Letters www.elsevier.com/locate/epsl Magma redox and structural controls on iron isotope variations in Earth’s mantle and crust ∗ N. Dauphas a, , M. Roskosz b, E.E. Alp c, D.R. Neuville d,M.Y.Huc,C.K.Sioa, F.L.H. Tissot a, J. Zhao c, L. Tissandier e,E.Médardf,C.Cordierb a Origins Laboratory, Department of the Geophysical Sciences and Enrico Fermi Institute, The University of Chicago, 5734 South Ellis Avenue, Chicago, IL 60637, USA b Unité Matériaux et Transformations, Université Lille 1, CNRS UMR 8207, 59655 Villeneuve d’Ascq, France c Advanced Photon Source, Argonne National Laboratory, 9700 South Cass Avenue, Argonne, IL 60439, USA d Géochimie et Cosmochimie, IPGP-CNRS, Sorbonne Paris Cité, 1 rue Jussieu, 75005 Paris, France e Centre de Recherches Pétrographiques et Géochimiques, CNRS-UPR 2300, 15 rue Notre-Dame des Pauvres, BP20, 54501 Vandoeuvre-lès-Nancy, France f Laboratoire Magmas et Volcans, Université Blaise Pascal, CNRS, IRD, 5 rue Kessler, 63038 Clermont-Ferrand, France article info abstract Article history: The heavy iron isotopic composition of Earth’s crust relative to chondrites has been explained by Received 13 July 2013 vaporization during the Moon-forming impact, equilibrium partitioning between metal and silicate at Received in revised form 15 April 2014 core–mantle-boundary conditions, or partial melting and magma differentiation. The latter view is Accepted 21 April 2014 supported by the observed difference in the iron isotopic compositions of MORBS and peridotites. -
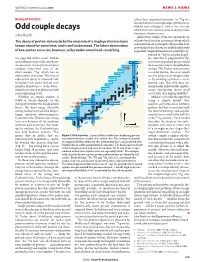
Odd Couple Decays Which Both One- and Two-Proton Decay Modes Juha Äystö Have Been Shown to Exist
19.1 N&V 273 MH 13/1/06 5:21 PM Page 279 NATURE|Vol 439|19 January 2006 NEWS & VIEWS NUCLEAR PHYSICS rather than sequential emission. As 94Ag was already known to undergo single-proton decay, Mukha and colleagues’ state is the first for Odd couple decays which both one- and two-proton decay modes Juha Äystö have been shown to exist. All previous studies of the two-proton decay The decay of proton-rich nuclei by the emission of a single proton has been of short-lived nuclear resonances from which known about for some time, and is well understood. The latest observation sequential decay is energetically possible have proved that these decays are indeed exclusively of two-proton emission, however, will provoke some head-scratching. sequential. Sequential emission is probably sup- pressed in 94Ag because the daugh- On page 298 of this issue1, Mukha ter state that is populated by the and colleagues report the simultane- Sn 50 first of two sequential decays would ous emission of two protons from a be an excited state of the palladium complex, long-lived state of the 94Ag isotope 93Pd. Rather than emitting silver isotope 94Ag, which has an a second proton, this state would odd number of protons. This type of tend to decay electromagnetically radioactive decay is expected only — by emitting a photon — to its for proton-rich nuclei with an even ground state. But why does the number of protons — so the obser- energetically unfavourable simulta- vation leaves nuclear physicists with neous two-proton decay itself some explaining to do. -
![Arxiv:1710.04254V5 [Astro-Ph.SR] 25 Jun 2019 Hr Xssawd Iest Nsei Se](https://docslib.b-cdn.net/cover/6203/arxiv-1710-04254v5-astro-ph-sr-25-jun-2019-hr-xssawd-iest-nsei-se-1946203.webp)
Arxiv:1710.04254V5 [Astro-Ph.SR] 25 Jun 2019 Hr Xssawd Iest Nsei Se
Accepted for publication in Astrophysical Journal Supplement, submitted on 19 September 2017, revised on 16 April 2018 Typeset using LATEX twocolumn style in AASTeX62 Explosive Nucleosynthesis in Near-Chandrasekhar Mass White Dwarf Models for Type Ia supernovae: Dependence on Model Parameters Shing-Chi Leung, Ken’ichi Nomoto1 1Kavli Institute for the Physics and Mathematics of the Universe (WPI), The University of Tokyo Institutes for Advanced Study The University of Tokyo, Kashiwa, Chiba 277-8583, Japan ABSTRACT We present two-dimensional hydrodynamics simulations of near-Chandrasekhar mass white dwarf (WD) models for Type Ia supernovae (SNe Ia) using the turbulent deflagration model with deflagration- detonation transition (DDT). We perform a parameter survey for 41 models to study the effects of the initial central density (i.e., WD mass), metallicity, flame shape, DDT criteria, and turbulent flame formula for a much wider parameter space than earlier studies. The final isotopic abundances of 11C to 91Tc in these simulations are obtained by post-process nucleosynthesis calculations. The survey includes SNe Ia models with the central density from 5 108 g cm−3 to 5 109 g cm−3 (WD masses × × of 1.30 - 1.38 M⊙), metallicity from 0 to 5 Z⊙, C/O mass ratio from 0.3 - 1.0 and ignition kernels including centered and off-centered ignition kernels. We present the yield tables of stable isotopes from 12C to 70Zn as well as the major radioactive isotopes for 33 models. Observational abundances of 55Mn, 56Fe, 57Fe and 58Ni obtained from the solar composition, well-observed SNe Ia and SN Ia remnants are used to constrain the explosion models and the supernova progenitor. -

High Specific Radioactivities of Cobalt, Platinum and Iridium From
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1951 High specific ar dioactivities of cobalt, platinum and iridium from photonuclear reactions Darleane Christian Iowa State College Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Physical Chemistry Commons Recommended Citation Christian, Darleane, "High specific ar dioactivities of cobalt, platinum and iridium from photonuclear reactions " (1951). Retrospective Theses and Dissertations. 13387. https://lib.dr.iastate.edu/rtd/13387 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. NOTE TO USERS This reproduction is the best copy available. UMI HIGH SPECIFIC MDIOACTIVITI OF COBALT, PLATIirOl-I Mm IRIDIUIl FROM PHOTOirUCL:mR R3ACTI0KS by Darloana Christian A Dissertation S-ubmitted to the Graduate Faculty in Partial Fulfillmant of The Requiramants for the Degrea of DOCTOR OP PHILOflOPIir Major Subject: Physical Chamistry Approveds Signature was redacted for privacy. In Cha3?i¥ of tlay6r' Worls: Signature was redacted for privacy. Heaa of tiajor Departraent ^ Signature was redacted for privacy. Daan of Graduate Collage loi/a State College 1951 UMI Number: DP12638 INFORMATION TO USERS The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleed-through, substandard margins, and improper alignment can adversely affect reproduction. -

Iron Isotope Systematics
Reviews in Mineralogy & Geochemistry Vol. 82 pp. XXX-XXX, 2017 11 Copyright © Mineralogical Society of America Iron Isotope Systematics Nicolas Dauphas Origins Lab Department of the Geophysical Sciences and Enrico Fermi Institute The University of Chicago 5734 South Ellis Avenue Chicago IL 60637 USA [email protected] Seth G. John University of Southern California Department of Earth Science Marine Trace Element Laboratory 3651 Trousdale Pkwy Los Angeles, CA 90089 USA [email protected] Olivier Rouxel IFREMER Department of Physical Resources and Deep-Sea Ecosystems Plouzané 29280 France University of Hawaii Department of Oceanography 1000 Pope Road Honolulu, HI 96822 USA [email protected] INTRODUCTION Iron is a ubiquitous element with a rich (i.e., complex) chemical behavior. It possesses three oxidation states, metallic iron (Fe0), ferrous iron (Fe2+) and ferric iron (Fe3+). The distribution of these oxidation states is markedly stratified in the Earth. • Metallic iron is primarily present in the core, where it is alloyed with Ni, Co, and light elements such as S, Si or O. Some metallic iron may be present at depth in the mantle because Fe2+ in bridgmanite can disproportionate into Fe3+ and Fe0 (Frost et al. 2004). Natural metallic iron also exists at the surface of the Earth in rare occurrences in the form of meteorite falls, metallic iron produced by reduction of lavas through interaction with coal sediments as in Disko Island (Greenland), and Josephinite (awaruite) produced by serpentinization reaction in peridotites. 1529-6466/17/0082-0011$10.00 http://dx.doi.org/10.2138/rmg.2017.82.11 Iron Isotope Systematics 277 • The main repository of ferrous iron is the mantle, where it is present in two spin states (the manner in which electrons fill the orbitals). -

High Precision Iron Isotope Measurements of Terrestrial and Lunar Materials
Geochimica et Cosmochimica Acta, Vol. 63, No. 11/12, pp. 1653–1660, 1999 Copyright © 1999 Elsevier Science Ltd Pergamon Printed in the USA. All rights reserved 0016-7037/99 $20.00 ϩ .00 PII S0016-7037(99)00089-7 High precision iron isotope measurements of terrestrial and lunar materials BRIAN L. BEARD* and CLARK M. JOHNSON Department of Geology and Geophysics, University of Wisconsin-Madison, Madison, Wisconsin 53706, USA (Received August 5, 1998; accepted in revised form February 11, 1999) Abstract—We present the analytical methods that have been developed for the first high-precision Fe isotope analyses that clearly identify naturally-occurring, mass-dependent isotope fractionation. A double-spike approach is used, which allows rigorous correction of instrumental mass fractionation. Based on 21 analyses of an ultra pure Fe standard, the external precision (1-SD) for measuring the isotopic composition of Fe is Ϯ0.14 ‰/mass; for demonstrated reproducibility on samples, this precision exceeds by at least an order of magnitude that of previous attempts to empirically control instrumentally-produced mass fractionation (Dixon et al., 1993). Using the double-spike method, 15 terrestrial igneous rocks that range in composition from peridotite to rhyolite, 5 high-Ti lunar basalts, 5 Fe-Mn nodules, and a banded iron formation have been analyzed for their iron isotopic composition. The terrestrial and lunar igneous rocks have the same isotopic compositions as the ultra pure Fe standard, providing a reference Fe isotope composition for the Earth and Moon. In contrast, Fe-Mn nodules and a sample of a banded iron formation have iron isotope compositions that vary over a relatively wide range, from ␦56Fe ϭϩ0.9 to Ϫ1.2 ‰; this range is 15 times the analytical errors of our technique. -

1 Excitation Functions and Isotopic Effects In
1 Excitation functions and isotopic effects in (n,p) reactions for stable iron isotopes from reaction threshold to 20 MeV. J. Joseph Jeremiah a, Damewan Suchiang b, B.M. Jyrwa a,* a Department of Physics, North Eastern Hill University, Shillong-793022, Meghalaya, India b Department of Physics, Tura Government College, Tura-794001, Meghalaya, India. * Corresponding Author. Email address : [email protected] (B.M. Jyrwa) Abstract The excitation functions for (n,p) reactions from reaction threshold to 20 MeV on four stable Iron isotopes viz; 54Fe,56Fe,57Fe and 58Fe were calculated using Talys-1.2 nuclear model code, this essentially involves fitting a set of global parameters. An excellent agreement between the calculated and experimental data is obtained with minimal effort on parameter fitting and only one free parameter called ‘Shell damping factor’ has been adjusted. This is very importance to the validation of nuclear model approaches with increased predictive power. The systematic decrease in (n, p) cross-sections with increasing neutron number in reactions induced by neutrons on isotopes of iron is explained in terms of the pre-equilibrium model. The compound nucleus and pre-equilibrium reaction mechanism as well as the isotopic effects were also studied extensively. Keywords Excitation functions, Shell damping factor, Compound nucleus model, Pre-equilibrium mechanism. 1. Introduction The common materials suitable for the reactor structures are stainless steel with Cr, Fe and Ni as main constituents. Structural materials are used to make the core and several other essential parts of a nuclear reactor. Thus, they must possess a high resistance to mechanical stress, stability to radiation and high temperature, and low neutron absorption.