Silicon Isotope Geochemistry: Fractionation Linked to Silicon Complexations and Its Geological Applications
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

12 Natural Isotopes of Elements Other Than H, C, O
12 NATURAL ISOTOPES OF ELEMENTS OTHER THAN H, C, O In this chapter we are dealing with the less common applications of natural isotopes. Our discussions will be restricted to their origin and isotopic abundances and the main characteristics. Only brief indications are given about possible applications. More details are presented in the other volumes of this series. A few isotopes are mentioned only briefly, as they are of little relevance to water studies. Based on their half-life, the isotopes concerned can be subdivided: 1) stable isotopes of some elements (He, Li, B, N, S, Cl), of which the abundance variations point to certain geochemical and hydrogeological processes, and which can be applied as tracers in the hydrological systems, 2) radioactive isotopes with half-lives exceeding the age of the universe (232Th, 235U, 238U), 3) radioactive isotopes with shorter half-lives, mainly daughter nuclides of the previous catagory of isotopes, 4) radioactive isotopes with shorter half-lives that are of cosmogenic origin, i.e. that are being produced in the atmosphere by interactions of cosmic radiation particles with atmospheric molecules (7Be, 10Be, 26Al, 32Si, 36Cl, 36Ar, 39Ar, 81Kr, 85Kr, 129I) (Lal and Peters, 1967). The isotopes can also be distinguished by their chemical characteristics: 1) the isotopes of noble gases (He, Ar, Kr) play an important role, because of their solubility in water and because of their chemically inert and thus conservative character. Table 12.1 gives the solubility values in water (data from Benson and Krause, 1976); the table also contains the atmospheric concentrations (Andrews, 1992: error in his Eq.4, where Ti/(T1) should read (Ti/T)1); 2) another category consists of the isotopes of elements that are only slightly soluble and have very low concentrations in water under moderate conditions (Be, Al). -

Atomic Mass of Silicon and Mcaco, 2
No. 4047 May 24, 1947 NATURE 705 18 LETTERS TO THE EDITORS p:• = 2·71036 gm.fcm.3 , d = 3·02945 A., and 1·09598. The Editors do not hold themselves responsible From the equations, for expressed by their correspondents. No u taken of anonymous communications Msio, = vaT N..4. • Psio,. a• . c Atomic Mass of Silicon and Mcaco, 2 . N A • Pcaco, . rp([3) • d'caco,, As is well known, the atomic mass of silicon has not yet been determined with satisfactory accuracy. and expressing the molecular mass of Si02 and Ca.C0 8 The value given in the International Table of Atomic as a function of the other magnitudes mentioned, Weigh.ts is 28·06. Practically the same value, 28·063, we conclude was g1ven by Baxter, Weatherill and Scriptural in Msio, = Mcaco, v'3 PstO, a• . c from the ratio SiC1 4/4Ag. But from the same -12 ' Pcaco, . rp([3) • d3caco,' ratiO, figures of 28·11 1 had been obtained in 1920 Substituting in this equation the various numerical bY:. .Weatherill :S:olmes• and of 28·106 by and Steinhell3 • Finally, in 1923, values, we find MsiO, = 60·075, the atomic mass of Weatherill and Brundage', from the ratio SiCl,fSi02, silicon being, therefore, gave the value 28·106 • The three of the four modern Si 28·075. gravimetric values are in fair agreement and lead This determination is based on the molecular mass to a m?an figure of 28·10 , which exceeds by 1/610 6 of calcite. This may not be the best reference sub approxrmately the figure given in the International stance for our purpose, since "it is well known to Table. -

The Silicon Isotope Composition of Ethmodiscus
The silicon isotope composition of Ethmodiscus rex laminated diatom mats from the tropical West Pacific: Implications for silicate cycling during the Last Glacial Maximum Zhifang Xiong, Tiegang Li, Thomas Algeo, Kristin Doering, Martin Frank, Mark Brzezinski, Fengming Chang, Sophie Opfergelt, Xavier Crosta, Fuqing Jiang, et al. To cite this version: Zhifang Xiong, Tiegang Li, Thomas Algeo, Kristin Doering, Martin Frank, et al.. The silicon isotope composition of Ethmodiscus rex laminated diatom mats from the tropical West Pacific: Implications for silicate cycling during the Last Glacial Maximum. Paleoceanography, American Geophysical Union, 2015, 30 (7), pp.803-823. 10.1002/2015PA002793. hal-02105557 HAL Id: hal-02105557 https://hal.archives-ouvertes.fr/hal-02105557 Submitted on 21 Apr 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. PUBLICATIONS Paleoceanography RESEARCH ARTICLE The silicon isotope composition of Ethmodiscus rex 10.1002/2015PA002793 laminated diatom mats from the tropical West Key Points: Pacific: Implications for silicate cycling • E. rex utilized dissolved silicon mainly from Asian dust during the LGM during the Last Glacial Maximum • Surface waters of the EPS were eutrophic for dissolved silicon during Zhifang Xiong1, Tiegang Li1, Thomas Algeo2,3, Kristin Doering4,5, Martin Frank5, Mark A. -

“Variation of Stable Silicon Isotopes: Analytical Developments and Applications in Precambrian Geochemistry”
“Variation of stable silicon isotopes: Analytical developments and applications in Precambrian geochemistry” Dissertation zur Erlangung des Grades “Doktor der Naturwissenschaften” am Fachbereich Chemie, Pharmazie und Geowissenschaften der Johannes Gutenberg-Universität in Mainz Kathrin Abraham geb.: 23.05.1980 in Nürnberg Mainz den, 30.03.2010 Dekan: 1. Berichterstatter: 2. Berichterstatter: Tag der mündlichen Prüfung: 28.05.2010 Preface This doctoral thesis was performed as cooperation between the University of Mainz and the Royal Museum for Central Africa, Belgium. Section I: Introduction Section II: δ30Si and δ29Si Determinations on USGS BHVO-1 and BHVO-2 Reference Materials with a New Configuration on a Nu Plasma Multi-Collector ICP-MS (Kathrin Abraham, Sophie Opfergelt, François Fripiat, Anne-Julie Cavagna, Jeroen T.M. de Jong., Stephen F. Foley, Luc André and Damien Cardinal) Geostandards and Geoanalytical Research, 2008. Section III: Coupled silicon-oxygen isotopic evidences for the origin of silicification in mafic volcanic rocks of the Barberton Greenstone Belt, South Africa (Kathrin Abraham, Axel Hofmann, Stephen F. Foley, Damien Cardinal, Chris Harris, Matthias Barth, Luc André) Submitted to Earth and Planetary Science Letters. Section IV: Potential changes in the source of Granitoids in the Archaean: the Si isotope perspective (Kathrin Abraham, Axel Hofmann, Stephen F. Foley, Damien Cardinal, Luc André) Submitted as extended Abstract, 5th International Archaean Symposium. In all cases, sample preparation, isotope (except of oxygen), trace element and further analytical work, data evaluation and interpretation as well as writing of the manuscripts were performed by the first author. Besides publications in peer-reviewed journals, results of the thesis were presented talks and poster presentations at several international conferences and workshops (Related publications). -

Isotopic Abundances and Atomic Weights of the Elements
Isotopic Abundances and Atomic Weights of the Elements Cite as: Journal of Physical and Chemical Reference Data 13, 809 (1984); https://doi.org/10.1063/1.555720 Published Online: 15 October 2009 Paul De Biévre, Marc Gallet, Norman E. Holden, and I. Lynus Barnes ARTICLES YOU MAY BE INTERESTED IN Isotopic Compositions of the Elements, 2001 Journal of Physical and Chemical Reference Data 34, 57 (2005); https:// doi.org/10.1063/1.1836764 Atomic Weights of the Elements 1999 Journal of Physical and Chemical Reference Data 30, 701 (2001); https:// doi.org/10.1063/1.1395055 Representative Equations for the Thermal Conductivity of Water Substance Journal of Physical and Chemical Reference Data 13, 893 (1984); https:// doi.org/10.1063/1.555718 Journal of Physical and Chemical Reference Data 13, 809 (1984); https://doi.org/10.1063/1.555720 13, 809 © 1984 American Institute of Physics for the National Institute of Standards and Technology. IsotopiC Abundances and Atomic Weights of the Elements Paul De Bifwre and Marc Gallet Central Bureau for Nuclear Measurements. Joint Research Centre. Commission of the European Communities. IT. S~ Geel. Belgium " C1 'il~ Norman E. Holden National Nuclear Data Center, Brookhayen National Laboratory, Upton. NY 11973. USA and I. Lynus Barnes National Measurement Laboratory, National Bureau of Standards. Gaithersburg, MD 20899, USA A large number of measurements describing the isotopic composition of the ele ments using a variety of analytical methods have been reported since the discovery of the first isotope in 1912. During the past several decades, however, mass spec trometric methods have been used, almost exclusively, to determine the isotopic composition, and thus the atomic weights, of the elements. -

HW 3.3 Pg 89 CH3R #7-23 Odd, #34
CSM Grade 10 Chemistry 2014-2015 HW 3.3 Pg 89 CH3R #7-23 odd, #34 7. a. What are isotopes? Atoms of the same element with different number of neutrons b. How are the isotopes of a particular element alike? They have the same number of protons and usually electrons depending on their charges. Also, their reaction behaviors or other properties are not significantly different than others. c. How are they different? They are different by the number of neutrons meaning their masses are different. Because the number of neutron really determine the stability of an atom, the abudance of each isotopes differ. 8. Copy and complete the following table concerning the three isotopes of silicon, Si. Isotope Number of protons Number of electrons Number of neutrons Si-28 14 14 14 Si-29 14 14 15 Si-30 14 14 16 9. a. What is the atomic number of an element? Atomic Number (Z) = number of protons b. What is the mass number of an isotope? Mass Number = number of protons + number of neutrons c. In the nuclear symbol for deuterium, , identify the atomic number and the mass number. 2 is the mass number and 1 is the atomic number. In general we have 11. Use the periodic table and the information that follows to write the hyphen notation for each isotope described. a. atomic number =2, mass number = 4; He-4 b. atomic number = 8, mass number = 16; O-16 CSM Grade 10 Chemistry 2014-2015 c. atomic number =19, mass number = 39; K-39 13. What is the atomic mass of an atom if its mass is approximately equal to the following? a. -

Stable Silicon Isotopes of Groundwater, Feldspars, and Clay Coatings in the Navajo Sandstone Aquifer, Black Mesa, Arizona, USA
Available online at www.sciencedirect.com Geochimica et Cosmochimica Acta 73 (2009) 2229–2241 www.elsevier.com/locate/gca Stable silicon isotopes of groundwater, feldspars, and clay coatings in the Navajo Sandstone aquifer, Black Mesa, Arizona, USA R.B. Georg a,c,*, C. Zhu b, B.C. Reynolds c, A.N. Halliday a a University of Oxford, Department of Earth Sciences, Parks Road, OX1 3PR Oxford, UK b Indiana University, Department of Geological Sciences, USA c ETH Zurich, Institute for Isotope Geochemistry & Mineral Resources, Switzerland Received 8 September 2008; accepted in revised form 3 February 2009; available online 14 February 2009 Abstract We present some of the first analyses of the stable isotopic composition of dissolved silicon (Si) in groundwater. The groundwater samples were from the Navajo Sandstone aquifer at Black Mesa, Arizona, USA, and the Si isotope composition of detrital feldspars and secondary clay coatings in the aquifer were also analyzed. Silicon isotope compositions were mea- sured using high-resolution multi-collector inductively coupled mass spectrometry (HR-MC-ICP-MS) (Nu1700 & NuPlasma HR). The quartz dominated bulk rock and feldspar separates have similar d30Si of À0.09 ± 0.04& and À0.15 ± 0.04& (±95% SEM), respectively, and clay separates are isotopically lighter by up to 0.4& compared to the feldspars. From isotopic mass- balance considerations, co-existing aqueous fluids should have d30Si values heavier than the primary silicates. Positive d30Si values were only found in the shallow aquifer, where Si isotopes are most likely fractionated during the dissolution of feld- spars and subsequent formation of clay minerals. -

Part A. Isotopic Abundance and the Mass Spectra of Elements 1
Name___________________________________________________ Part A. Isotopic Abundance and the Mass Spectra of Elements 1. The modern definition of isotopes is based on the subatomic particle structure of atoms. Complete the following table to show the number of protons, neutrons, and electrons in neutral atoms of Cl–35 and Cl–37. Isotope Cl–35 Cl–37 Protons Neutrons 18 20 Electrons 2. Write a 2−3 sentence definition for isotopes that includes all of the following terms: protons, neutrons, atomic number, and mass number. 3. Which property of an atom defines the identity of an element? Explain why isotopes form the same compounds and undergo the same reactions. 4. Radioactive isotopes (radioisotopes) are widely used in medicine. Because isotopes have identical chemical properties, the reaction and distribution of radioisotopes in the body is similar to that of their natural isotopes. Iodine–131, for example, is an artificial radioisotope that is used to diagnose thyroid disorders. When administered to a patient, I–131 is taken up by the thyroid gland, where it is incorporated into the thyroid hormone, just as iodine in the diet would be. Based on where the following elements are likely to be found in the body, match each radioisotope with its medical use. Sodium-24 a. studies of bone formation Phosphorus-32 b. red blood cell studies Calcium-47 c. tracing blood circulation Iron-55 d. genetics (DNA) research 5. Regardless of its source on earth, the element chlorine always contains 75.8% chlorine-35 atoms and 24.2% chlorine-37 atoms. The atomic mass of an element is the weighted average of the masses of the isotopes. -

A Review of the Stable Isotope Bio-Geochemistry of the Global Silicon Cycle and Its Associated Trace Elements Jill Sutton, Luc André, Damien Cardinal, Daniel J
A Review of the Stable Isotope Bio-geochemistry of the Global Silicon Cycle and Its Associated Trace Elements Jill Sutton, Luc André, Damien Cardinal, Daniel J. Conley, Gregory F. de Souza, Jonathan Dean, Justin Dodd, Claudia Ehlert, Michael Ellwood, Patrick Frings, et al. To cite this version: Jill Sutton, Luc André, Damien Cardinal, Daniel J. Conley, Gregory F. de Souza, et al.. A Review of the Stable Isotope Bio-geochemistry of the Global Silicon Cycle and Its Associated Trace Elements. Frontiers in Earth Science, Frontiers Media, 2018, 5, pp.UNSP 112. 10.3389/feart.2017.00112. hal- 01832229 HAL Id: hal-01832229 https://hal.archives-ouvertes.fr/hal-01832229 Submitted on 28 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. REVIEW published: 30 January 2018 doi: 10.3389/feart.2017.00112 A Review of the Stable Isotope Bio-geochemistry of the Global Silicon Cycle and Its Associated Trace Elements Jill N. Sutton 1*, Luc André 2, Damien Cardinal 3, Daniel J. Conley 4,5, Gregory F. de Souza 6, Jonathan Dean 7,8, Justin Dodd 9, Claudia Ehlert 10, Michael J. Ellwood 11, Patrick J. -

Non-Traditional Fractionation of Non-Traditional Isotopes: Evaporation, Chemical Diffusion and Soret Diffusion
Chemical Geology 258 (2009) 92–103 Contents lists available at ScienceDirect Chemical Geology journal homepage: www.elsevier.com/locate/chemgeo Non-traditional fractionation of non-traditional isotopes: Evaporation, chemical diffusion and Soret diffusion Frank M. Richter a,⁎, Nicolas Dauphas a,b, Fang-Zhen Teng a,1 a The University of Chicago, Department of the Geophysical Sciences, 5734 South Ellis Avenue, Chicago, IL 60637, United States b The University of Chicago, Enrico Fermi Institute, 5640 South Ellis Avenue, Chicago, IL 60637, United States article info abstract Article history: Recent developments documenting high temperature isotope fractionations of a variety of elements (Li, Mg, Accepted 5 June 2008 Si, Ca, Fe, Ni) by a variety of processes (evaporation, chemical diffusion and thermal diffusion) are reviewed along with some recent applications of these fractionations to Earth and meteoritic problems. Keywords: © 2008 Elsevier B.V. All rights reserved. Isotope fractionation Evaporation Chemical diffusion Thermal diffusion Grain boundary diffusion 1. Introduction (Teng et al., 2008a). This N 0.2‰ Fe isotopic variation is interpreted to be caused by fractional crystallization, with an estimated isotopic The study and application of stable isotope fractionations has a fractionation between minerals and melts of ~ − 0.1‰. The fractiona- long and distinguished history in chemical geology. An important tion of Fe isotopes at high temperature could be produced by kinetic or reason for this is that equilibrium fractionations are temperature -

Isotopic Engineering of Silicon by Jacoby Jackson B.S., Texas A&M
Isotopic engineering of silicon by Jacoby Jackson B.S., Texas A&M University, 2017 A REPORT Submitted in partial fulfillment of the requirements for the degree MASTER OF SCIENCE Tim Taylor Department of Chemical Engineering Carl R. Ice College of Engineering KANSAS STATE UNIVERSITY Manhattan, Kansas 2019 Approved by: Major Professor Dr. James Edgar Copyright © Jacoby Jackson 2019. Abstract Isotopically engineering silicon to achieve nearly 100 % pure 28Si crystals has been researched for use in metrology, quantum computing, and in silicon-based integrated circuits. Silicon is naturally composed of three isotopes: 28Si, 29Si, and 30Si with a relative abundance of 92.2%, 4.7%, and 3.1%, respectively. Use of isotope engineering of silicon was explored in the International Avogadro Project to define Avogadro’s constant. The silicon employed for this project must be as isotopically and chemically pure as possible to reduce defects in the crystal lattice. Isotope engineered silicon has also been researched for utilization in quantum computing. A proposal by B.E Kane has phosphorous donor atoms imbedded in silicon. The nuclear spin of the phosphorous atoms are the qubits for this scheme. Phosphorous has a nuclear spin (I=±1/2) and 28Si does not. Isotopically engineered silicon is needed for this scheme to reduce the concentration of 29Si, which also has a nuclear spin of I=±1/2 and is a major source of decoherence. The isotope composition of silicon also affects its phonon related properties. Thermal conductivity is of interest due to the increasing importance of heat management in integrated circuits. Isotopically engineered silicon has a thermal conductivity ten times higher than that of silicon of natural composition at 24 K and is 8% higher at room temperature. -
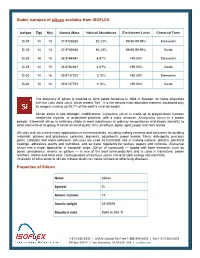
Stable Isotopes of Silicon Available from ISOFLEX Properties of Silicon
Stable isotopes of silicon available from ISOFLEX Isotope Z(p) N(n) Atomic Mass Natural Abundance Enrichment Level Chemical Form Si-28 14 14 27.9769265 92.23% 99.90-99.99% Elemental Si-28 14 14 27.9769265 92.23% 99.90-99.99% Oxide Si-29 14 15 28.9764947 4.67% >99.00% Elemental Si-29 14 15 28.9764947 4.67% >99.00% Oxide Si-30 14 16 29.9737702 3.10% >99.00% Elemental Si-30 14 16 29.9737702 3.10% >99.00% Oxide The discovery of silicon is credited to Jöns Jakob Berzelius in 1824 in Sweden. Its name originates with the Latin word silicis, which means “flint.” It is the second most abundant element, exceeded only by oxygen, making up 25.7% of the earth's crust by weight. Silicon exists in two allotropic modifications: Crystalline silicon is made up of grayish-black lustrous needle-like crystals, or octahedral platelets, with a cubic structure. Amorphous silicon is a brown powder. Elemental silicon is relatively stable in most substances at ordinary temperatures and shows similarity to other elements of its group. It exists as sand quartz, flint, amethyst, agate, opal, jasper and rock crystal. Silicates and silica have many applications in numerous fields, including making cements and concretes for building materials, glasses and glassware, ceramics, pigments, adsorbents, paper boards, fillers, detergents, precious gems, catalysts and water-softeners. Silicones are used as lubricants and in making rubbers, plastics, electrical coatings, adhesives, paints and varnishes, and as water repellents for textiles, papers and concrete. Elemental silicon has a major application in computer chips.