Structural and Functional Studies of Proteins of Medical Relevance
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Type of the Paper (Article
Supplementary Material A Proteomics Study on the Mechanism of Nutmeg-induced Hepatotoxicity Wei Xia 1, †, Zhipeng Cao 1, †, Xiaoyu Zhang 1 and Lina Gao 1,* 1 School of Forensic Medicine, China Medical University, Shenyang 110122, P. R. China; lessen- [email protected] (W.X.); [email protected] (Z.C.); [email protected] (X.Z.) † The authors contributed equally to this work. * Correspondence: [email protected] Figure S1. Table S1. Peptide fraction separation liquid chromatography elution gradient table. Time (min) Flow rate (mL/min) Mobile phase A (%) Mobile phase B (%) 0 1 97 3 10 1 95 5 30 1 80 20 48 1 60 40 50 1 50 50 53 1 30 70 54 1 0 100 1 Table 2. Liquid chromatography elution gradient table. Time (min) Flow rate (nL/min) Mobile phase A (%) Mobile phase B (%) 0 600 94 6 2 600 83 17 82 600 60 40 84 600 50 50 85 600 45 55 90 600 0 100 Table S3. The analysis parameter of Proteome Discoverer 2.2. Item Value Type of Quantification Reporter Quantification (TMT) Enzyme Trypsin Max.Missed Cleavage Sites 2 Precursor Mass Tolerance 10 ppm Fragment Mass Tolerance 0.02 Da Dynamic Modification Oxidation/+15.995 Da (M) and TMT /+229.163 Da (K,Y) N-Terminal Modification Acetyl/+42.011 Da (N-Terminal) and TMT /+229.163 Da (N-Terminal) Static Modification Carbamidomethyl/+57.021 Da (C) 2 Table S4. The DEPs between the low-dose group and the control group. Protein Gene Fold Change P value Trend mRNA H2-K1 0.380 0.010 down Glutamine synthetase 0.426 0.022 down Annexin Anxa6 0.447 0.032 down mRNA H2-D1 0.467 0.002 down Ribokinase Rbks 0.487 0.000 -

Folate Cycle Enzyme MTHFD1L Confers Metabolic Advantages in Hepatocellular Carcinoma
RESEARCH ARTICLE The Journal of Clinical Investigation Folate cycle enzyme MTHFD1L confers metabolic advantages in hepatocellular carcinoma Derek Lee,1 Iris Ming-Jing Xu,1 David Kung-Chun Chiu,1 Robin Kit-Ho Lai,1 Aki Pui-Wah Tse,1 Lynna Lan Li,1 Cheuk-Ting Law,1 Felice Ho-Ching Tsang,1 Larry Lai Wei,1 Cerise Yuen-Ki Chan,1 Chun-Ming Wong,1,2 Irene Oi-Lin Ng,1,2 and Carmen Chak-Lui Wong1,2 1Department of Pathology and 2State Key Laboratory for Liver Research, The University of Hong Kong, Hong Kong, China. Cancer cells preferentially utilize glucose and glutamine, which provide macromolecules and antioxidants that sustain rapid cell division. Metabolic reprogramming in cancer drives an increased glycolytic rate that supports maximal production of these nutrients. The folate cycle, through transfer of a carbon unit between tetrahydrofolate and its derivatives in the cytoplasmic and mitochondrial compartments, produces other metabolites that are essential for cell growth, including nucleotides, methionine, and the antioxidant NADPH. Here, using hepatocellular carcinoma (HCC) as a cancer model, we have observed a reduction in growth rate upon withdrawal of folate. We found that an enzyme in the folate cycle, methylenetetrahydrofolate dehydrogenase 1–like (MTHFD1L), plays an essential role in support of cancer growth. We determined that MTHFD1L is transcriptionally activated by NRF2, a master regulator of redox homeostasis. Our observations further suggest that MTHFD1L contributes to the production and accumulation of NADPH to levels that are sufficient to combat oxidative stress in cancer cells. The elevation of oxidative stress through MTHFD1L knockdown or the use of methotrexate, an antifolate drug, sensitizes cancer cells to sorafenib, a targeted therapy for HCC. -

Differential Requirements for Tousled-Like Kinases 1 and 2 in Mammalian Development
Cell Death and Differentiation (2017) 24, 1872–1885 & 2017 Macmillan Publishers Limited, part of Springer Nature. All rights reserved 1350-9047/17 www.nature.com/cdd Differential requirements for Tousled-like kinases 1 and 2 in mammalian development Sandra Segura-Bayona1,8, Philip A Knobel1,8, Helena González-Burón1,8, Sameh A Youssef2,3, Aida Peña-Blanco1, Étienne Coyaud4,5, Teresa López-Rovira1, Katrin Rein1, Lluís Palenzuela1, Julien Colombelli1, Stephen Forrow1, Brian Raught4,5, Anja Groth6, Alain de Bruin2,7 and Travis H Stracker*,1 The regulation of chromatin structure is critical for a wide range of essential cellular processes. The Tousled-like kinases, TLK1 and TLK2, regulate ASF1, a histone H3/H4 chaperone, and likely other substrates, and their activity has been implicated in transcription, DNA replication, DNA repair, RNA interference, cell cycle progression, viral latency, chromosome segregation and mitosis. However, little is known about the functions of TLK activity in vivo or the relative functions of the highly similar TLK1 and TLK2 in any cell type. To begin to address this, we have generated Tlk1- and Tlk2-deficient mice. We found that while TLK1 was dispensable for murine viability, TLK2 loss led to late embryonic lethality because of placental failure. TLK2 was required for normal trophoblast differentiation and the phosphorylation of ASF1 was reduced in placentas lacking TLK2. Conditional bypass of the placental phenotype allowed the generation of apparently healthy Tlk2-deficient mice, while only the depletion of both TLK1 and TLK2 led to extensive genomic instability, indicating that both activities contribute to genome maintenance. Our data identifies a specific role for TLK2 in placental function during mammalian development and suggests that TLK1 and TLK2 have largely redundant roles in genome maintenance. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

S42003-019-0587-Z.Pdf
Corrected: Author Correction ARTICLE https://doi.org/10.1038/s42003-019-0587-z OPEN High-resolution crystal structure of human asparagine synthetase enables analysis of inhibitor binding and selectivity Wen Zhu 1,10, Ashish Radadiya 1, Claudine Bisson2,10, Sabine Wenzel 3, Brian E. Nordin4,11, 1234567890():,; Francisco Martínez-Márquez3, Tsuyoshi Imasaki 3,5, Svetlana E. Sedelnikova2, Adriana Coricello 1,6,7, Patrick Baumann 1, Alexandria H. Berry8, Tyzoon K. Nomanbhoy4, John W. Kozarich 4, Yi Jin 1, David W. Rice 2, Yuichiro Takagi 3 & Nigel G.J. Richards 1,9 Expression of human asparagine synthetase (ASNS) promotes metastatic progression and tumor cell invasiveness in colorectal and breast cancer, presumably by altering cellular levels of L-asparagine. Human ASNS is therefore emerging as a bona fide drug target for cancer therapy. Here we show that a slow-onset, tight binding inhibitor, which exhibits nanomolar affinity for human ASNS in vitro, exhibits excellent selectivity at 10 μM concentration in HCT- 116 cell lysates with almost no off-target binding. The high-resolution (1.85 Å) crystal structure of human ASNS has enabled us to identify a cluster of negatively charged side chains in the synthetase domain that plays a key role in inhibitor binding. Comparing this structure with those of evolutionarily related AMP-forming enzymes provides insights into intermolecular interactions that give rise to the observed binding selectivity. Our findings demonstrate the feasibility of developing second generation human ASNS inhibitors as lead compounds for the discovery of drugs against metastasis. 1 School of Chemistry, Cardiff University, Cardiff, UK. 2 Department of Molecular Biology and Biotechnology, University of Sheffield, Sheffield, UK. -

Sarcoma Metabolomics: Current Horizons and Future Perspectives
cells Review Sarcoma Metabolomics: Current Horizons and Future Perspectives Miguel Esperança-Martins 1,2,3,* , Isabel Fernandes 1,3,4 , Joaquim Soares do Brito 4,5, Daniela Macedo 6, Hugo Vasques 4,7, Teresa Serafim 2, Luís Costa 1,3,4 and Sérgio Dias 2,4 1 Centro Hospitalar Universitário Lisboa Norte, Medical Oncology Department, Hospital Santa Maria, 1649-028 Lisboa, Portugal; [email protected] (I.F.); [email protected] (L.C.) 2 Vascular Biology & Cancer Microenvironment Lab, Instituto de Medicina Molecular João Lobo Antunes, Faculdade de Medicina, Universidade de Lisboa, 1649-028 Lisboa, Portugal; tserafi[email protected] (T.S.); [email protected] (S.D.) 3 Translational Oncobiology Lab, Instituto de Medicina Molecular João Lobo Antunes, Faculdade de Medicina, Universidade de Lisboa, 1649-028 Lisboa, Portugal 4 Faculdade de Medicina, Universidade de Lisboa, 1649-028 Lisboa, Portugal; [email protected] (J.S.d.B.); [email protected] (H.V.) 5 Centro Hospitalar Universitário Lisboa Norte, Orthopedics and Traumatology Department, Hospital Santa Maria, 1649-028 Lisboa, Portugal 6 Medical Oncology Department, Hospital Lusíadas Lisboa, 1500-458 Lisboa, Portugal; [email protected] 7 General Surgery Department, Instituto Português de Oncologia de Lisboa Francisco Gentil, 1099-023 Lisboa, Portugal * Correspondence: [email protected] Abstract: The vast array of metabolic adaptations that cancer cells are capable of assuming, not Citation: Esperança-Martins, M.; only support their biosynthetic activity, but also fulfill their bioenergetic demands and keep their Fernandes, I.; Soares do Brito, J.; intracellular reduction–oxidation (redox) balance. Spotlight has recently been placed on the en- Macedo, D.; Vasques, H.; Serafim, T.; ergy metabolism reprogramming strategies employed by cancer cells to proliferate. -

Anti-MTHFD1 Monoclonal Antibody (DCABH- 12465) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-MTHFD1 monoclonal antibody (DCABH- 12465) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Antigen Description This gene encodes a protein that possesses three distinct enzymatic activities, 5,10- methylenetetrahydrofolate dehydrogenase, 5,10-methenyltetrahydrofolate cyclohydrolase and 10-formyltetrahydrofolate synthetase. Each of these activities catalyzes one of three sequential reactions in the interconversion of 1-carbon derivatives of tetrahydrofolate, which are substrates for methionine, thymidylate, and de novo purine syntheses. The trifunctional enzymatic activities are conferred by two major domains, an aminoterminal portion containing the dehydrogenase and cyclohydrolase activities and a larger synthetase domain. Immunogen A synthetic peptide of human MTHFD1 is used for rabbit immunization. Isotype IgG Source/Host Rabbit Species Reactivity Human Purification Protein A Conjugate Unconjugated Applications Western Blot (Transfected lysate); ELISA Size 1 ea Buffer In 1x PBS, pH 7.4 Preservative None Storage Store at -20°C or lower. Aliquot to avoid repeated freezing and thawing. GENE INFORMATION Gene Name MTHFD1 methylenetetrahydrofolate dehydrogenase (NADP+ dependent) 1, methenyltetrahydrofolate cyclohydrolase, formyltetrahydrofolate synthetase [ Homo sapiens ] Official Symbol MTHFD1 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved Synonyms MTHFD1; methylenetetrahydrofolate -
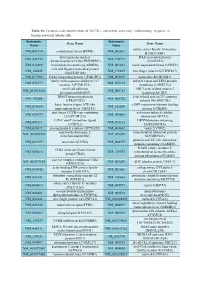
Systematic Name Gene Name Systematic Name Gene Name NM 001710 Complement Factor B(CFB) NM 052831 Solute Carrier Family 18 Member
Table S1: Genome-wide identification of SGLT2i`s interaction with early inflammatory response in human proximal tubular cells. Systematic Systematic Gene Name Gene Name Name Name solute carrier family 18 member NM_001710 complement factor B(CFB) NM_052831 B1(SLC18B1) heterogeneous nuclear DAZ associated protein NM_031372 NM_170711 ribonucleoprotein D like(HNRNPDL) 1(DAZAP1) NM_014299 bromodomain containing 4(BRD4) NM_001261 cyclin dependent kinase 9(CDK9) cilia and flagella associated protein NM_182628 NM_178835 zinc finger protein 827(ZNF827) 100(CFAP100) NM_017906 PAK1 interacting protein 1(PAK1IP1) NM_024015 homeobox B4(HOXB4) family with sequence similarity 167 ankyrin repeat and LEM domain NM_053279 NM_015114 member A(FAM167A) containing 2(ANKLE2) small cell adhesion ARP3 actin related protein 3 NM_001031628 NM_005721 glycoprotein(SMAGP) homolog(ACTR3) TRAF3 interacting protein actin related protein 2/3 complex NM_147686 NM_005720 2(TRAF3IP2) subunit 1B(ARPC1B) basic leucine zipper ATF-like cAMP responsive element binding NM_018664 NM_182898 transcription factor 3(BATF3) protein 5(CREB5) zinc finger CCCH-type containing activation induced cytidine NM_025079 NM_020661 12A(ZC3H12A) deaminase(AICDA) C-X-C motif chemokine ligand DENN domain containing NM_001511 NM_015213 1(CXCL1) 5A(DENND5A) NM_025072 prostaglandin E synthase 2(PTGES2) NM_004665 vanin 2(VNN2) superoxide dismutase 2, mitochondrial ribosomal protein NM_001024465 NM_016070 mitochondrial(SOD2) S23(MRPS23) jumonji and AT-rich interaction NM_033199 urocortin 2(UCN2) NM_004973 -

MTHFD1 Is a Genetic Interactor of BRD4 and Links Folate Metabolism to Transcriptional Regulation
bioRxiv preprint doi: https://doi.org/10.1101/439422; this version posted October 10, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. MTHFD1 is a genetic interactor of BRD4 and links folate metabolism to transcriptional regulation Sara Sdelci1, André F. Rendeiro1, Philipp Rathert2,3, Gerald Hofstätter1,4, Anna Ringler1,4, Herwig P. Moll5, Wanhui You1,4, Kristaps Klavins1, Bettina Gürtl1, Matthias Farlik1, Sandra Schick1,4, Freya Klepsch1, Matthew Oldach1, Pisanu Buphamalai1, Fiorella Schischlik1, Peter Májek1, Katja Parapatics1, Christian Schmidl1, Michael Schuster1, Thomas Penz1, Dennis L. Buckley6, Otto Hudecz7, Richard Imre7, Robert Kralovics1, Keiryn L. Bennett1, Andre C. Müller1, Karl Mechtler2, Jörg Menche1, James E. Bradner6, Georg E. Winter1, Emilio Casanova5,8, Christoph Bock1,9,10, Johannes Zuber2, Stefan Kubicek1,4,* Affiliations: 1CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences. Lazarettgasse 14, 1090 Vienna, Austria. 2Research Institute of Molecular Pathology (IMP), Vienna Biocenter (VBC), 1030 Vienna, Austria. 3Current address: Institute for Biochemistry (IBC), University Stuttgart, 70569 Stuttgart, Germany. 4Christian Doppler Laboratory for Chemical Epigenetics and Antiinfectives, CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences, Vienna, Austria. 5Department of Physiology, Center of Physiology and Pharmacology & Comprehensive Cancer Center (CCC), Medical University of Vienna, Vienna, Austria 6Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA 02215 7Institute of Molecular Biotechnology, Austrian Academy of Sciences, Vienna Biocenter (VBC), 1030 Vienna, Austria. 8Ludwig Boltzmann Institute for Cancer Research (LBI-CR), Vienna, Austria 9Department of Laboratory Medicine, Medical University of Vienna, 1090 Vienna, Austria. -

Molecular Signatures Differentiate Immune States in Type 1 Diabetes Families
Page 1 of 65 Diabetes Molecular signatures differentiate immune states in Type 1 diabetes families Yi-Guang Chen1, Susanne M. Cabrera1, Shuang Jia1, Mary L. Kaldunski1, Joanna Kramer1, Sami Cheong2, Rhonda Geoffrey1, Mark F. Roethle1, Jeffrey E. Woodliff3, Carla J. Greenbaum4, Xujing Wang5, and Martin J. Hessner1 1The Max McGee National Research Center for Juvenile Diabetes, Children's Research Institute of Children's Hospital of Wisconsin, and Department of Pediatrics at the Medical College of Wisconsin Milwaukee, WI 53226, USA. 2The Department of Mathematical Sciences, University of Wisconsin-Milwaukee, Milwaukee, WI 53211, USA. 3Flow Cytometry & Cell Separation Facility, Bindley Bioscience Center, Purdue University, West Lafayette, IN 47907, USA. 4Diabetes Research Program, Benaroya Research Institute, Seattle, WA, 98101, USA. 5Systems Biology Center, the National Heart, Lung, and Blood Institute, the National Institutes of Health, Bethesda, MD 20824, USA. Corresponding author: Martin J. Hessner, Ph.D., The Department of Pediatrics, The Medical College of Wisconsin, Milwaukee, WI 53226, USA Tel: 011-1-414-955-4496; Fax: 011-1-414-955-6663; E-mail: [email protected]. Running title: Innate Inflammation in T1D Families Word count: 3999 Number of Tables: 1 Number of Figures: 7 1 For Peer Review Only Diabetes Publish Ahead of Print, published online April 23, 2014 Diabetes Page 2 of 65 ABSTRACT Mechanisms associated with Type 1 diabetes (T1D) development remain incompletely defined. Employing a sensitive array-based bioassay where patient plasma is used to induce transcriptional responses in healthy leukocytes, we previously reported disease-specific, partially IL-1 dependent, signatures associated with pre and recent onset (RO) T1D relative to unrelated healthy controls (uHC). -

Differentially Expressed Genes Yuki
supplementary Table 1. The differentially expressed genes in F3-treated THP-1 cells after 6 hours. Gene Symbol Chip ID Fold Change Genbank UniGene Description CCL20 205476_at 421.7 NM_004591 Hs.75498 chemokine (C-C motif) ligand 20 CCL4 204103_at 351.6 NM_002984 Hs.75703 chemokine (C-C motif) ligand 4 BCL2A1 205681_at 228.6 NM_004049 Hs.227817 BCL2-related protein A1 ISG20 204698_at 202.1 NM_002201 Hs.459265 interferon stimulated exonuclease gene 20kDa CCL8 214038_at 139.1 AI984980 Hs.271387 chemokine (C-C motif) ligand 8 CXCL1 204470_at 115.1 NM_001511 Hs.789 chemokine (C-X-C motif) ligand 1 (melanoma growth stimulating activity, alpha) IL7R 205798_at 111.6 NM_002185 Hs.362807 interleukin 7 receptor ; interleukin 7 receptor BCL3 204908_s_at 104.2 NM_005178 Hs.31210 B-cell CLL/lymphoma 3 GBP1 202269_x_at 99.99 BC002666 Hs.62661 guanylate binding protein 1, interferon-inducible, 67kDa SLAMF7 219159_s_at 98.3 NM_021181 Hs.517265 SLAM family member 7 CXCL10 204533_at 93.05 NM_001565 Hs.413924 chemokine (C-X-C motif) ligand 10 IL8 211506_s_at 91.31 AF043337 Hs.624 interleukin 8 IL8 202859_x_at 85.26 NM_000584 Hs.624 interleukin 8 THBS1 201110_s_at 84.5 NM_003246 Hs.164226 thrombospondin 1 EBI3 219424_at 82.87 NM_005755 Hs.501452 Epstein-Barr virus induced gene 3 SAT 213988_s_at 76.47 BE971383 Hs.28491 spermidine/spermine N1-acetyltransferase USP18 219211_at 76.22 NM_017414 Hs.38260 ubiquitin specific peptidase 18 LAMP3 205569_at 75.89 NM_014398 Hs.518448 lysosomal-associated membrane protein 3 CCL2 216598_s_at 71.33 S69738 Hs.303649 chemokine