Chapter 4: Detailed Human Health Risk Assessment
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Creighton-Flin Flon
Flin Flon Domain of Saskatchewan – Dataset Descriptions The Flin Flon domain hosts one of the most prolific Precambrian volcanogenic massive sulphide (VMS) districts in the world, with over 160 million tonnes of ore produced from at least 28 deposits in Saskatchewan and Manitoba since the start of the 20th century. Situated in the Reindeer Zone of the of the Trans-Hudson Orogen, rocks of this domain comprise dominantly metamorphosed and polydeformed volcanoplutonic terranes with subordinate siliciclastic sedimentary sequences. Volcanic rocks vary in composition throughout the domain and were originally emplaced in a variety of tectonic settings, including island arc(s), arc rift, ocean floor, and ocean pleateau. The VMS deposits are hosted primarily by Paleoproterozoic juvenile arc rocks in one of several defined lithotectonic assemblages. The datasets provided for this area are located within a ~12,000 km2 area of the Flin Flon Domain of Saskatchewan that contains several past producing base metal mines and a multitude of known mineral occurrences. The northern quarter of the area is underlain by exposed Precambrian Shield, whereas the southern three-quarters consists of Precambrian basement situated beneath up to 200 metres of undeformed, Phanerozoic sedimentary rocks. The sedimentary cover in this southern portion makes exploration of the Precambrian rocks particularly challenging. The datasets provided for this area consist of: (i) airborne geophysical survey data, and (ii) multiple GIS datasets from the exposed Precambrian Shield and/or the buried Precambrian basement and/or the Phanerozoic sedimentary cover. The geophysical data comprises digital data for 22 airborne surveys including industry-derived data submitted to the Saskatchewan Geological Survey through assessment work reports, and surveys funded by provincial and federal governments. -

Pictographs in Northern Saskatchewan: Vision Quest
PICTOGRAPHS IN NORTHERN SASKATCHEWAN: VISION QUEST AND PAWAKAN A Thesis Submitted to the Faculty of Graduate Studies and Research in Partial Fulfillment of the Requirements for a Degree of Master of Arts in the Department of Anthropology and Archaeology University of Saskatchewan Saskatoon by Katherine A. Lipsett April, 1990 The author claims copyright. Use shall not be made of the material contained herein without proper acknowledgement, as indicated on the following page. The author has agreed that the Library, University of Saskatchewan, may make this thesis freely available for inspection. Moreover, the author has agreed that permission for extensive copying of this thesis for scholarly purposes may be granted by the professor or professors who supervised the thesis work recorded herein or, in their absence, by the Head of the Department or the Dean of the College in which the thesis work was done. It is understood that due recognition will be given to the author of this thesis and to the University of Saskatchewan in any use of the material in this thesis. Copying or publication or any other use of the thesis for financial gain without approval by the University of Saskatchewan and the author's written permission is prohibited. Requests for permission to copy or to make any other use of material in this thesis in whole or part should be addressed to: Head of the Department of Anthropology and Archaeology University of Saskatchewan Saskatoon, Saskatchewan Canada S7N OWO i ABSTRACT Pictographs in northern Saskatchewan have been linked to the vision quest ritual by Rocky Cree informants. -

New Results from Mapping in the Flin Flon Mining Camp, Creighton, Saskatchewan
Stratigraphy, Structure, and Silicification: New Results From Mapping in the Flin Flon Mining Camp, Creighton, Saskatchewan Kate MacLachlan MacLachlan (2006): Stratigraphy, structure, and silicification: new results from mapping in the Flin Flon Mining Camp, Creighton, Saskatchewan; in Summary of Investigations Volume 2, 2006, Saskatchewan Geological Survey, Sask. Industry Resources, Misc. Rep. 2006-4.2, CD-ROM, Paper A-9, 25p. Abstract Two areas in the Flin Flon mining camp were mapped at a scale of 1:5000, one area between Douglas Lake and West Arm Road on the west limb of the Beaver Road Anticline, and the other on the peninsula between Green Lake and Phantom Lake. In the Douglas Lake area, mapping was undertaken in the hanging wall of the west-younging, felsic-dominated Myo member of the Flin Flon formation. Significant findings include, an extrusive felsic unit above the upper contact of the Myo member, abundant peperite at several stratigraphic levels, an up to 100 m wide zone of semi-conformable silicification, and pervasive sinistral shearing that post-dates the Phantom Intrusive Suite. In the Green Lake Peninsula area, three fault-bounded successions with opposing younging directions were recognized. The easternmost succession, east of the newly defined Burley Lake Fault (a modified version of the Phantom Lake Fault), is part of the west-younging Louis formation on the east limb of the Burley Lake Syncline. The east-younging succession between the Burley Lake and Green Lake faults comprises a large component of fine- grained mafic volcaniclastic rocks intercalated with aphyric and plagioclase ± pyroxene porphyritic flows, and is tentatively correlated with the Hidden formation. -

Geology and Geochemistry of the Schist Lake Mine Area, Flin Flon, Manitoba (Part of NTS 63K12) by E.M Cole1, S.J
GS-2 Geology and geochemistry of the Schist Lake mine area, Flin Flon, Manitoba (part of NTS 63K12) by E.M Cole1, S.J. Piercey1,2 and H.L. Gibson1 Cole, E.M., Piercey, S.J. and Gibson, H.L. 2008: Geology and geochemistry of the Schist Lake mine area, Flin Flon, Manitoba (part of NTS 63K12); in Report of Activities 2008, Manitoba Science, Technology, Energy and Mines, Manitoba Geological Survey, p. 18–28. Summary regarding the stratigraphic and The goal of this two-year research project is to temporal relationship between the characterize and describe the Schist Lake and Mandy main deposits — Callinan, Triple volcanogenic massive sulphide (VMS) deposits and their 7 and Flin Flon — and the Schist hostrocks, and to determine if these VMS deposits are Lake–Mandy deposits located just a few kilometres to the time-stratigraphic equivalent to those occurring in the south of the main camp. The objective of this research main Flin Flon camp. The first year of this project focused is to document the volcanic and structural setting of the on the area around the Schist Lake mine, where detailed Schist Lake and Mandy deposits, document the alteration lithofacies and alteration facies maps were completed, types associated with these deposits, and ultimately along with detailed field descriptions of each lithofacies. compare the Schist Lake and Mandy deposits with the The mine is hosted by a succession of mafic and felsic aforementioned current VMS deposits of the main Flin volcaniclastic rocks that were deposited in a basin envi- Flon camp. ronment. Geochemical -

History of Mining in Saskatchewan
History of Mining In Saskatchewan Early Mining in Saskatchewan The earliest mining occurred when earth’s inhabitants started using various stones for tools or certain clays for cooking vessels. The earliest recorded occupation in Saskatchewan was around 9000 B.C. at the Niska site in the southern part of the province. Ample evidence of the use of stone tools, arrow heads, and spear heads, etc. has been found in the area. Much of the material used by these early inhabitants was imported or traded from other regions of North America. The study of the stone tools provides us with information about the people’s work, their history, their religion, their travels and their relationships with other groups or nations. Stone is readily available throughout most of Saskatchewan. This was especially important for Saskatchewan’s First Nations people who moved their camps frequently in search for food. The stones available were not all suitable for tools and they needed a constant supply of stone material that broke cleanly or was hard enough for pounding. Consequently, they made regular trips to the source areas or traded with people who lived near the sources. For these early residents of our province, the exchange of goods was more than just a means of acquiring things. Bartering and gift exchange was a means of creating and reinforcing relationships between individuals, families and nations. For thousands of years, goods have been exchanged through networks that extended across North America. Although perishable goods were also traded, our records are in the form of shell or stone artefacts. -

Human Health Risk Assessment for Flin Flon, Manitoba and Creighton, Saskatchewan
Independent Expert Review Panel (IERP) Human Health Risk Assessment for Flin Flon, Manitoba and Creighton, Saskatchewan Volume II June 23-24, 2009 Winnipeg, CA Peer Consultation Organized by: Toxicology Excellence for Risk Assessment (http://www.tera.org/peer/) August 28, 2009 Peer Consultation Meeting on the Relationship Between Polycyclic Aromatic Compound (PAC) Profile and Toxicity of Petroleum Substances This page intentionally left blank. Independent Expert Review Panel (IERP) Human Health Risk Assessment for Flin Flon, Manitoba and Creighton, Saskatchewan 2 Table of Contents Appendix A ............................................................................................................... 1 Conflict of Interest ................................................................................................................... A3 Biographical Sketches of Panel Members ............................................................................... A5 Ronald Brecher, Ph.D., C.Chem., QPRA, DABT ............................................................... A5 Michael L. Dourson, Ph.D., DABT, FATS ......................................................................... A6 Susan Griffin, Ph.D., DABT ................................................................................................ A7 Sean Hays, M.S. ................................................................................................................... A8 Norm Healey, B.Sc., DABT ............................................................................................... -

From the Bush to the Village in Northern Saskatchewan: Contrasting CCF Community Development Projects"
CORE Metadata, citation and similar papers at core.ac.uk Provided by Érudit Article "From the Bush to the Village in Northern Saskatchewan: Contrasting CCF Community Development Projects" David M. Quiring Journal of the Canadian Historical Association / Revue de la Société historique du Canada, vol. 17, n° 1, 2006, p. 151-178. Pour citer cet article, utiliser l'information suivante : URI: http://id.erudit.org/iderudit/016106ar DOI: 10.7202/016106ar Note : les règles d'écriture des références bibliographiques peuvent varier selon les différents domaines du savoir. Ce document est protégé par la loi sur le droit d'auteur. L'utilisation des services d'Érudit (y compris la reproduction) est assujettie à sa politique d'utilisation que vous pouvez consulter à l'URI https://apropos.erudit.org/fr/usagers/politique-dutilisation/ Érudit est un consortium interuniversitaire sans but lucratif composé de l'Université de Montréal, l'Université Laval et l'Université du Québec à Montréal. Il a pour mission la promotion et la valorisation de la recherche. Érudit offre des services d'édition numérique de documents scientifiques depuis 1998. Pour communiquer avec les responsables d'Érudit : [email protected] Document téléchargé le 9 février 2017 10:21 From the Bush to the Village in Northern Saskatchewan: Contrasting CCF Community Development Projects DAVID M. QUIRING Abstract The election of the CCF in 1944 brought rapid change for the residents of northern Saskatchewan. CCF initiatives included encouraging northern aboriginals to trade their semi-nomadic lifestyles for lives in urban settings. The establishment of Kinoosao on Reindeer Lake provides an example of how CCF planners established new villages; community development processes excluded local people. -

Wildnest-Tabbernor Transect: Mirond-Pelican Lakes Area (Parts of NTS 63M-2 and -3)1
Wildnest-Tabbernor Transect: Mirond-Pelican Lakes Area (Parts of NTS 63M-2 and -3)1 K.E. Ashton and R. Shi 2 Ashton, K.E. and Shi, R. (1994): Wildnest-Tabbernor Transect: Mirond-Pelican lakes area (parts of NTS 63M-2 and -3); in Sum mary of Investigations 1994, Saskatchewan Geological Survey, Sask. Energy Mines, Misc. Rep. 94-4. Mapping along the Wildnest-Tabbernor Transect Previous detailed mapping in the area {Macdonald, {Ashton and Leclair, 1991 ) was extended westward in 1974; Macdonald and MacQuarrie, 1978; Sibba'd, 1994 to incorporate the Mirond-Pelican lakes area. The 1978) has been followed up by recent studies directed study covered a 240 km2 area of the northwestern Han toward elucidating the tectonic history of the area son Lake Block centred 12 km south of Pelican Nar {Lewry and Macdonald, 1988; Lewry et al., 1989; Craig, rows and 75 km northwest of Flin Flon. Manitoba 1989; Shi and Lewry, 1991 , 1992; Sun et al., 1991 , (Figure 1). The area is accessible from Highway 135 1992, 1993). Seismic reflection data has been collected which links the villages of Pelican Narrows and Sandy along an east-west line 30 km to the south {Lucas et Bay to the Hanson Lake Road {Highway 106). al., 1993; Lewry et al., 1994) and on a north-south line along Highway 135 (Lucas et al., 1994) as part of the LITHOPROBE Trans-Hudson Orogen Transect. ATT ITTI KISSEYNEW z <( L TRANSECT 0 0 WILONCST - ATT ITll LAKES AR EA .. · w 1991 z- z w _J G BLOCK KM FLON DOMAIN ~·· 45' Figure 1 - Location of the Wildnest-Tabbemor Transect (dots) showing completed work. -
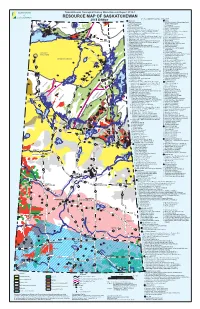
Mineral Resource Map of Saskatchewan
Saskatchewan Geological Survey Miscellaneous Report 2018-1 RESOURCE MAP OF SASKATCHEWAN KEY TO NUMBERED MINERAL DEPOSITS† 2018 Edition # URANIUM # GOLD NOLAN # # 1. Laird Island prospect 1. Box mine (closed), Athona deposit and Tazin Lake 1 Scott 4 2. Nesbitt Lake prospect Frontier Adit prospect # 2 Lake 3. 2. ELA prospect TALTSON 1 # Arty Lake deposit 2# 4. Pitch-ore mine (closed) 3. Pine Channel prospects # #3 3 TRAIN ZEMLAK 1 7 6 # DODGE ENNADAI 5. Beta Gamma mine (closed) 4. Nirdac Creek prospect 5# # #2 4# # # 8 4# 6. Eldorado HAB mine (closed) and Baska prospect 5. Ithingo Lake deposit # # # 9 BEAVERLODGE 7. 6. Twin Zone and Wedge Lake deposits URANIUM 11 # # # 6 Eldorado Eagle mine (closed) and ABC deposit CITY 13 #19# 8. National Explorations and Eldorado Dubyna mines 7. Golden Heart deposit # 15# 12 ### # 5 22 18 16 # TANTATO # (closed) and Strike deposit 8. EP and Komis mines (closed) 14 1 20 #23 # 10 1 4# 24 # 9. Eldorado Verna, Ace-Fay, Nesbitt Labine (Eagle-Ace) 9. Corner Lake deposit 2 # 5 26 # 10. Tower East and Memorial deposits 17 # ###3 # 25 and Beaverlodge mines and Bolger open pit (closed) Lake Athabasca 21 3 2 10. Martin Lake mine (closed) 11. Birch Crossing deposits Fond du Lac # Black STONY Lake 11. Rix-Athabasca, Smitty, Leonard, Cinch and Cayzor 12. Jojay deposit RAPIDS MUDJATIK Athabasca mines (closed); St. Michael prospect 13. Star Lake mine (closed) # 27 53 12. Lorado mine (closed) 14. Jolu and Decade mines (closed) 13. Black Bay/Murmac Bay mine (closed) 15. Jasper mine (closed) Fond du Lac River 14. -

Manitoba's Precambrian Drillcore Libraries Program
GS-22 Manitoba’s Precambrian Drillcore Libraries Program by D.E. Prouse Prouse, D.E. 2007: Manitoba’s Precambrian Drillcore Libraries Program; in Report of Activities 2007, Manitoba Science, Technology, Energy and Mines, Manitoba Geological Survey, p. 211. Summary 143 Main Street, Suite 201 Manitoba’s Mineral Resources Division has been Flin Flon, Manitoba R8A 1K2 storing Precambrian drillcore, obtained primarily from Telephone: (204) 687-1632 exploration drilling, since the early 1970s. Since that time, E-mail: [email protected] the Manitoba government has created a substantial reposi- Access to view core at the tory of drillcore at five locations throughout the province. Brady Road facility in Winnipeg Funding to help build a significant inventory of drillcore should be arranged with to represent crucial stratigraphy within Manitoba’s princi- Jim Payne, Assessment Geologist pal mining districts has been sporadic since the collection Mines Branch was started. Since 2001, funding has allowed the depart- Manitoba Science, Technology, Energy and Mines ment primarily to maintain drillcore inventories at current 1395 Ellice Avenue, Suite 360 levels, with some additions being made to the Thompson Winnipeg, Manitoba, R3G 3P2 and Lynn Lake facilities. Telephone: (204) 945-6535 Email: [email protected] Introduction Once permission has been granted to view noncon- The Manitoba Mineral Resources Division considers fidential core in a specific library, arrangements will be the archiving of exploration drillcore to be a valuable data made for the user to obtain keys to that facility. Keys for source for use by mineral-exploration companies and access to the Centennial compound near Flin Flon can researchers. -

Northern Saskatchewan Arts and Culture Handbook
Northern Saskatchewan Arts and Culture Handbook Compiled and edited by Miriam Körner for the Northern Sport, Culture and Recreation District Acknowledgements First and foremost I would like to thank the Northern Sport, Culture and Recreation District for contracting me to develop the Northern Saskatchewan Arts and Culture Handbook. Through this work I had the rare opportunity to visit many of Northern Saskatchewan’s finest arts and crafts people. I would like to thank all the artists that invited me into their homes, and shared their stories TheThe Northern Sport, Culture & with me. It was a wonderful experience and there is just one regret: That I did not have more RecreationRecreation DistrictDistrict wwouldould liklikee to time to spend with all of you! thankthank theirtheir sponsors:sponsors: This project would not have been possible without the outstanding support through dedicated individuals that helped me along my journey through northern communities. I especially would like to thank Rose Campbell & the Ospwakun Sepe School in Brabant; Simon Bird and Mary Thomas & the Reindeer Lake School in Southend; Marlene Naldzil & the Fond du Lac Denesuline First Nation Band Office, Rita Adam & the Father Gamache Memorial School, Flora Augier for room and board; Raymond McDonald and Corinne Sayazie & the Black Lake Denesuline Nation Band Office with special thanks to Mary Jane Cook for a great job finding artists, translating and lots of fun! Thanks to the Peter Ballantyne Cree Nation Band Office in Pelican Narrows and special thanks to Georgina Linklater for driving as well as Sheldon Dorion and Gertie Dorion for translating. Too bad I couldn’t stay longer! In Sandy Bay I would like to thank Lorraine Bear & the Hector Thiboutot Community School. -

Island Falls, Saskatchewan: 1929-1967
Island Falls, Saskatchewan: 1929-1967 Island Falls, Saskatchewan: 1929-1967 Introduction Contents Guestbook News http://www.islandfalls.ca/[02/01/2015 8:35:26 AM] Introduction to Island Falls, Saskatchewan Island Falls, Saskatchewan: 1929 - 1967 INTRODUCTION You can find Island Falls on the map of Saskatchewan at 55.5º N, 102.4º W, about sixty miles by airplane north of Flin Flon, Manitoba, just west of the Manitoba border. Nowadays it is the name of a SaskPower hydro electric power station. However, the Island Falls to which this website is dedicated was a small settlement of about two hundred people serving the power plant. Located on a man-made island in the Churchill River near the power plant, the settlement was called "The Camp" by its residents, the families of the operators, electricians, machinists, administrators, labourers, and men of many other skills employed by the CRP Co. (Churchill River Power Company), a subsidiary of the HBM&S Co. (Hudson Bay Mining and Smelting Company). The Island Falls power plant was built between 1928 and 1930 to provide electricity for the HBM&S copper and zinc mining and smelting operation in Flin Flon. Thirty-seven years later, when technology permitted the power plant to be run by remote control, management decided the residential settlement was no longer needed. As a result of "Automation" in 1967, Island Falls employees and their families moved to positions in Flin Flon or into retirement. Afterwards, the company houses, which were very modern for their time, remained vacant but intact until the winter of 1971 when the electricity that heated them was interrupted.