208352Orig1s000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
![United States Patent 1191 Lll] 3,983,266 Bahls [451 Sept](https://docslib.b-cdn.net/cover/8181/united-states-patent-1191-lll-3-983-266-bahls-451-sept-158181.webp)
United States Patent 1191 Lll] 3,983,266 Bahls [451 Sept
United States Patent 1191 lll] 3,983,266 Bahls [451 Sept. 28, 1976 [54] METHOD FOR APPLYING METALLIC 3.776.740 [2/1973 Sivcrz ct al. .......................... .. l06/l SILVER TO A SUBSTRATE OTHER PUBLICATIONS [75] Inventor: Harry Bahls, Wayne, Pa. lvanov et al., Chem. Abs. 43:2548c, I949, [73] Assignee: Peacock Laboratories, Inc., Philadelphia, Pa. Primary Examiner-Ralph S. Kendall [22] Filed: Oct. 9, I974 I 57] ABSTRACT [2!] ,Appl. No.: 513,417 High efficiency deposition of silver on the surface of a substrate is obtained by providing a solution contain [52] [1.8. CI ............................... .. 427/164; 427/165; ing reducible dissolved silver in the presence of an al 427/168; 427/424; 427/426; l06/l kali metal hydroxide and ammonia, all of which are [51] Int. CLZ. ......................................... .. C23C 3/02 applied to the substrate in the presence of an aqueous [58] Field of Search .............. .. l06/l; 427/l68. I69, solution of a moderating reducer containing :1 poly 427/165, I64, 426, 304, 125, 425 hydric alcohol of the formula CH2OH(CHOH),,C H,OH, where n is an integer from 1 to 6. Preferably [56] References Cited the polyhydric alcohol is sorbitol, and in a preferred UNITED STATES PATENTS embodiment a moderator is the form of a thio glycerol is present. 2,996,406 8/l96l Weinrich ........... ............ .. 427/168 3,772,078 ll/l973 Polichcttc ct al ................. .. l06/l X l5 Claims, No Drawings 3,983,266 1 Other objects and advantages of this invention, in METHOD FOR APPLYING METALLIC SILVER TO cluding the economy of the same, and the case with A SUBSTRATE which it may be applied to existing silver coating equip ment and apparatus, will further become apparent BRIEF SUMMARY OF THE INVENTION 5 hereinafter. -

Biochemistry Lab (BT35L) Lab Manual
Department of Biotechnology Sri Jayachamarajendra College of Engineering Mysuru-570006 Biochemistry Lab (BT35L) Lab Manual (Prepared by Dr. S. Nanjunda Swamy and Dr. M. N. Nagendra Prasad) INDEX 1. PREPARATION OF SODIUM ACETATE BUFFER 2. ESTIMATION OF GLUCOSE BY DNS METHOD 3. ESTIMATION OF SAPONIFICATION VALUE OF FATS/OILS 4. ESTIMATION OF ACID VALUE OF FATS/OILS 5. PREPARATION OF PHOSPHOTIDYL CHOLINE FROM EGG YOLK 6. LOWRY METHOD FOR PROTEIN ESTIMATION 7. ESTIMATION OF AMINO ACID BY FORMAL TITRATION 8. DETERMINATION OF ISOELECTRIC POINT OF GLYCINE (PI) 9. SEPARATION OF AMINO ACIDS BY CIRCLULAR PAPER CHROMATOGRAPHY 10. ESTIMATION OF IRON BY WONG’S METHOD Experiment No 1: PREPARATION OF SODIUM ACETATE BUFFER Aim: Preparation of sodium acetate buffer of 0.1M and 4.7 pH (25ml) Principle: Buffer is a solution used to maintain the pH of any biochemical reaction. Procedure: 1. Weigh about 0.205g of sodium acetate. Dissolve it in 15ml distilled water. 2. Dip the electrode of the pre calibrated pH meter in the above solution. 3. Add acetic acid of 0.5M drop by drop till the pH reaches 4.7. 4. Make up the solution to 25ml with distilled water. 5. Store the buffer in a sealed container. Experiment No 2: ESTIMATION OF GLUCOSE BY DNS METHOD Aim: To estimate the amount of glucose in the given ample by DNS method. Principle: This method tests for the presence of free carbonyl group (C=O), of the reducing sugars. The reaction involves the oxidation of this of free carbonyl group (aldehyde functional group present in, glucose and the ketone functional group in fructose) and simultaneous reduction of , 3,5-dinitrosalicylic acid to 3-amino,5- nitrosalicylic acid (a coloured molecule) under alkaline conditions. -

Disposal of Solid Chemicals in the Normal Trash
Disposal of Solid Chemicals in the Normal Trash Many solid chemicals can be safety discarded into the normal trash, provided they are in containers that are not broken or cracked and have tightly fitting caps. These chemicals are considered acceptable for ordinary disposal because they display none of the properties of hazardous waste, are of low acute toxicity, and have not been identified as having any chronic toxic effects as summarized in the National Institute of Occupational Safety and Health (NIOSH) “Registry of Toxic Effects of Chemical Substances”. Examples of chemicals acceptable for disposal as regular trash are listed below. To dispose of these chemicals, place the containers in a box lined with a plastic bag, tape the top of the box shut, write “Normal Trash” on the box and then place the box next to the lab trash container. Only solid forms of these chemicals can be disposed in this manner. Any questions about these chemicals or other chemicals that may be disposed of in the normal trash should be directed to the Hazardous Materials Technician (610) 330-5225. Chemicals Generally Acceptable for Disposal as Regular Trash Acacia powder, gum Detergent (most) Methyl salicylate Sodium carbonate arabic Cation exchange resins Methylene blue Sodium chloride Acid, Ascorbic Chromatographic Methyl stearate Sodium citrate Acid, Benzoic absorbents Nutrient agar Sodium dodecyl sulfate Acid, Boric Crystal violet Octacosane (SDS) Acid, Casamind Dextrin Parafin Sodium formate Acid, Citric Dextrose Pepsin Sodium iodide Acid, Lactic Diatomaceous -

Dietary Supplements Compendium Volume 1
2015 Dietary Supplements Compendium DSC Volume 1 General Notices and Requirements USP–NF General Chapters USP–NF Dietary Supplement Monographs USP–NF Excipient Monographs FCC General Provisions FCC Monographs FCC Identity Standards FCC Appendices Reagents, Indicators, and Solutions Reference Tables DSC217M_DSCVol1_Title_2015-01_V3.indd 1 2/2/15 12:18 PM 2 Notice and Warning Concerning U.S. Patent or Trademark Rights The inclusion in the USP Dietary Supplements Compendium of a monograph on any dietary supplement in respect to which patent or trademark rights may exist shall not be deemed, and is not intended as, a grant of, or authority to exercise, any right or privilege protected by such patent or trademark. All such rights and privileges are vested in the patent or trademark owner, and no other person may exercise the same without express permission, authority, or license secured from such patent or trademark owner. Concerning Use of the USP Dietary Supplements Compendium Attention is called to the fact that USP Dietary Supplements Compendium text is fully copyrighted. Authors and others wishing to use portions of the text should request permission to do so from the Legal Department of the United States Pharmacopeial Convention. Copyright © 2015 The United States Pharmacopeial Convention ISBN: 978-1-936424-41-2 12601 Twinbrook Parkway, Rockville, MD 20852 All rights reserved. DSC Contents iii Contents USP Dietary Supplements Compendium Volume 1 Volume 2 Members . v. Preface . v Mission and Preface . 1 Dietary Supplements Admission Evaluations . 1. General Notices and Requirements . 9 USP Dietary Supplement Verification Program . .205 USP–NF General Chapters . 25 Dietary Supplements Regulatory USP–NF Dietary Supplement Monographs . -

Official Journal of the European Communities No L 61/1
18 . 3 . 95 EN Official Journal of the European Communities No L 61/1 I (Acts whose publication is obligatory) EUROPEAN PARLIAMENT AND COUNCIL DIRECTIVE No 95/2/EC of 20 February 1995 on food additives other than colours and sweeteners THE EUROPEAN PARLIAMENT AND THE COUNCIL OF Whereas it is necessary to lay down strict rules for the THE EUROPEAN UNION, use of food additives in infant formulae, follow-on formulae and weaning foods, as referred to in Council Having regard to the Treaty establishing the European Directive 89/398/EEC of 3 May 1989 on the Community, and in particular Article 100a thereof, approximation of the laws of the Member States relating to foodstuffs intended for particular nutritional uses ( 5 ), Having regard to the proposal from the Commission ( 1 ), and in particular Article 4(1 ) ( e ) thereof; Having regard to the opinion of the Economic and Social Whereas this Directive is not intended to affect rules Committee ( 2 ), relating to sweeteners and colours; Acting in accordance with the procedure laid down in Whereas, pending specific provisions pursuant to Council Article 189b of the Treaty ( 3 ), Directive 91/414/EEC of 15 July 1991 concerning the placing of plant protection products on the market ( 6 ), Having regard to the Council Directive 89/107/EEC of and pursuant to Council Directive 90/642/EEC of 21 December 1988 on the approximation of the laws of 27 November 1990 on the fixing of maximum levels for the Member States concerning food additives authorized pesticide residues in and on certain products of plant -

Potassium Sodium Tartrate MSDS # 592.00
Material Safety Data Sheet Page 1 of 2 Potassium Sodium Tartrate MSDS # 592.00 Section 1: Product and Company Identification Potassium Sodium Tartrate Synonyms/General Names: Rochelle Salt. Product Use: For educational use only Manufacturer: Columbus Chemical Industries, Inc., Columbus, WI 53925. 24 Hour Emergency Information Telephone Numbers CHEMTREC (USA): 800-424-9300 CANUTEC (Canada): 613-424-6666 ScholAR Chemistry; 5100 W. Henrietta Rd, Rochester, NY 14586; (866) 260-0501; www.Scholarchemistry.com Section 2: Hazards Identification White crystalline powder, no odor. HMIS (0 to 4) Health 0 This material is not considered hazardous. Fire Hazard 0 Target organs: None known. Reactivity 0 This material is not considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200) if used properly. Section 3: Composition / Information on Ingredients Potassium Sodium Tartrate (6381-59-5), >99% Section 4: First Aid Measures Always seek professional medical attention after first aid measures are provided. Eyes: Immediately flush eyes with excess water for 15 minutes, lifting lower and upper eyelids occasionally. Skin: Immediately flush skin with excess water for 15 minutes while removing contaminated clothing. Ingestion: Call Poison Control immediately. Rinse mouth with cold water. Give victim 1-2 cups of water or milk to drink. Induce vomiting immediately. Inhalation: Remove to fresh air. If not breathing, give artificial respiration. Section 5: Fire Fighting Measures When heated to decomposition, emits acrid fumes. 0 Protective equipment and precautions for firefighters: Use foam or dry chemical to extinguish fire. 0 0 Firefighters should wear full fire fighting turn-out gear and respiratory protection (SCBA). Cool container with water spray. -

Non-Hazardous Chemicals
OSU HAZARDOUS WASTE PROCESS DETERMINATIONM F ORRevision 10/12/2018 Written records are required to demonstrate compliance for all wastes, both hazardous and non-hazardous, that may reasonably be suspected of being hazardous waste. If you have any questions, please contact EH&S at (541)737-2273 or email us at [email protected] Please submit the completed Or save it and email to email address above. Generation Location: Generator Name: (generator any person, whose act or process produces a hazardous waste) Building: Room: Process Name: Contact Phone Number: Generation Process: (how was the waste created) Estimated Volume of Waste Generation: (per month)__________________________________________ mL Liter Gal other_______________ Attachments: (attach all applicable documentation describing the waste (e.g. Acceptable Generator Knowledge, SDS, SOP etc.) : List all hazardous constituents, the volume must equal 100% Chemical: Volume % Compound: Volume % 1 5. 2 6. 3 7. 4. 8. Physical State: Characteristics: (Select all that apply) Solid Flashpoint _________________ Solid w/freestanding or absorbed liquid pH __________________ Liquid (if liquid, indicate if the liquid is: Reactive to Air Single – Phase Reactive to Water Multi - Phase Reactive to pressure Other: ___________________ Oxidizer Toxic Radioactive Biological 1 OSU CHEMICAL WASTE PROCESS DETERMINATION FORM Draft Revision 01/23/19 This section is to be filled out by EH&S. Process: NOTE: • This determination must be reviewed annually and whenever there are process or raw material changes that could affect the waste. • This written documentation of Hazardous Waste Process Determination is required by Federal regulation to be retained until 3 years AFTER the waste is no longer being accumulated. -

Sodium L(+)-Tartrate
SODIUM L(+)-TARTRATE Prepared at the 7th JECFA (1963), published in NMRS 35 (1964) and in FNP 52 (1992). Metal and arsenic specifications revised at the 63rd JECFA (2004) (identical to those listed for potassium sodium L(+)-tartrate). An ADI of 0-30 mg/kg bw was established at the 17th JECFA (1973) and reconfirmed at the 21st JECFA (1977). SYNONYMS Sodium dextro-tartrate; INS No. 335 DEFINITION Chemical names Disodium L-tartrate, disodium (+)-tartrate, disodium (+)-2,3- dihydroxybutanedioic acid C.A.S. number 868-18-8 Chemical formula C4H4Na2O6 · 2H2O Structural formula Formula weight 230.8 Assay Not less than 99% after drying DESCRIPTION Transparent, colourless and odourless crystals FUNCTIONAL USES Sequestrant and stabilizer in meat products and sausage casings CHARACTERISTICS IDENTIFICATION Solubility (Vol. 4) One gram is soluble in 3 ml of water; insoluble to ethanol Test for tartrate (Vol. 4) Passes test Test for sodium (Vol. 4) Passes test PURITY Loss on drying (Vol. 4) Not more than 17% and not less than 14% (150o, 3 h) pH (Vol. 4) 7.0 - 7.5 (1 in 10 soln) Oxalate Add 5 drops of dilute acetic acid TS and 2 ml of calcium chloride TS to 10 ml of a 10% solution of sodium tartrate. No turbidity is produced within 1 h. Lead (Vol. 4) Not more than 2 mg/kg Determine using an atomic absorption technique appropriate to the specified level. The selection of sample size and method of sample preparation may be based on the principles of the method described in Volume 4, “Instrumental Methods.” METHOD OF Weigh 1.500 g of the dried sample into a tared porcelain crucible, ignite ASSAY gently at first, until the salt is thoroughly carbonized, protecting the carbonized salt from contact with the flame at all times. -

Solid Chemicals for the Normal Trash
SOLID CHEMICALS FOR THE NORMAL TRASH Solid chemicals acceptable for disposal as regular trash are listed below: Acacia powder, gum arabic Calcium silicate Manganese sulfate Acid, Ascorbic Calcium sulfate Methyl red Acid, Benzoic Detergent (most) Methyl salicylate Acid, Boric Cation exchange resins Methylene blue Acid, Casamind Crystal violet Methyl stearate Acid, Citric Dextrin Nutrient agar Acid, Lactic Dextrose Octacosane Acid, Oleic Diatomaceous earth Parafin Acid, Phthalic Docosanoic acid Pepsin Acid, Salicycle Drierite (calcium sulfate, anhydrous) Peptone Acid, Silicic Ferric oxide Petroleum jelly Acid, Stearic Ferric phosphate Polyethylene, solid Acid, Succinic Ferric pyrophosphate Polystryene Acid, Tartaric Ferric sulfate Potassium acetate Acrylamide gels Ferrous ammonium sulfate Potassium bicarbonate Agar(s) Galactose Potassium bromide Albumen Geletin Potassium carbonate Alumina Gum arabic Potassium chloride Aluminum oxide Gum guaiac Potassium citrate Amino acids, naturally occurring Hexadecanol, 1- Potassium ferricyanide Ammonium bicarbonate Kaolin Potassium iodide Ammonium phosphate Lactose Potassium phosphate Ammonium sulfate Lanolin Potassium sodium tartrate Ammonium sulfamate Lauric acid Potassium sulfate Base, blood agar Lauryl sulfate Potassium sulfite Beef extract Lithium carbonate Potassium sulfocyanate Behenic acid Lithium chloride Pumice Bentonite Lithium sulfate Salts, naturally occurring Brain heart infusion Litmus Sand Bromphenol blue Magnesium carbonate Silica Broth, nutrient Magnesium chloride Silica gel, unused -

EUROPEAN PHARMACOPOEIA Free Access to Supportive Pharmacopoeial Texts in the Field of Vaccines for Human Use During the Coronavirus Disease (COVID-19) Pandemic
EUROPEAN PHARMACOPOEIA Free access to supportive pharmacopoeial texts in the field of vaccines for human use during the coronavirus disease (COVID-19) pandemic Updated package - October 2020 Published in accordance with the Convention on the Elaboration of a European Pharmacopoeia (European Treaty Series No. 50) European Directorate Direction européenne for the Quality de la qualité of Medicines du médicament & HealthCare & soins de santé Council of Europe Strasbourg Free access to supportive pharmacopoeial texts in the field of vaccines for human use during the coronavirus disease (COVID-19) pandemic Updated package The EDQM is committed to supporting users during the coronavirus disease (COVID-19) pandemic – as well as contributing to the wider global effort to combat the virus – by openly sharing knowledge and providing access to relevant guidance/standards. To support organisations involved in the development, manufacture or testing of COVID-19 vaccines worldwide, many of which are universities and small and medium-sized enterprises, the EDQM is offeringte mporary free access to texts of the European Pharmacopoeia (Ph. Eur.) in the field of vaccines. This package includes quality standards for vaccines which developers can take into account to help build appropriate analytical control strategies for their COVID-19 candidate vaccines and ensure the quality and safety of the final product. Application of such quality requirements may ultimately help to facilitate regulatory acceptance of a subsequent marketing authorisation application. For ease of reading, a summary table listing the pharmacopoeial texts, with information regarding the vaccine types or vaccine platforms concerned (e.g. live attenuated viral vaccine, recombinant viral-vectored vaccines) is provided. -

Chemical Compatibility Storage Group
CHEMICAL SEGREGATION Chemicals are to be segregated into 11 different categories depending on the compatibility of that chemical with other chemicals The Storage Groups are as follows: Group A – Compatible Organic Acids Group B – Compatible Pyrophoric & Water Reactive Materials Group C – Compatible Inorganic Bases Group D – Compatible Organic Acids Group E – Compatible Oxidizers including Peroxides Group F– Compatible Inorganic Acids not including Oxidizers or Combustible Group G – Not Intrinsically Reactive or Flammable or Combustible Group J* – Poison Compressed Gases Group K* – Compatible Explosive or other highly Unstable Material Group L – Non-Reactive Flammable and Combustible, including solvents Group X* – Incompatible with ALL other storage groups The following is a list of chemicals and their compatibility storage codes. This is not a complete list of chemicals, but is provided to give examples of each storage group: Storage Group A 94‐75‐7 2,4‐D (2,4‐Dichlorophenoxyacetic acid) 94‐82‐6 2,4‐DB 609-99-4 3,5-Dinitrosalicylic acid 64‐19‐7 Acetic acid (Flammable liquid @ 102°F avoid alcohols, Amines, ox agents see SDS) 631-61-8 Acetic acid, Ammonium salt (Ammonium acetate) 108-24-7 Acetic anhydride (Flammable liquid @102°F avoid alcohols see SDS) 79‐10‐7 Acrylic acid Peroxide Former 65‐85‐0 Benzoic acid 98‐07‐7 Benzotrichloride 98‐88‐4 Benzoyl chloride 107-92-6 Butyric Acid 115‐28‐6 Chlorendic acid 79‐11‐8 Chloroacetic acid 627‐11‐2 Chloroethyl chloroformate 77‐92‐9 Citric acid 5949-29-1 Citric acid monohydrate 57-00-1 Creatine 20624-25-3 -
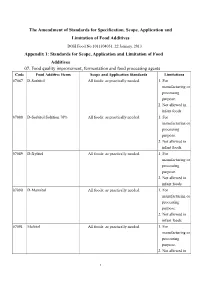
The Amendment of Standards for Specification, Scope, Application
The Amendment of Standards for Specification, Scope, Application and Limitation of Food Additives DOH Food No.1011304051, 22 January, 2013 Appendix 1: Standards for Scope, Application and Limitation of Food Additives 07. Food quality improvement, fermentation and food processing agents Code Food Additive Items Scope and Application Standards Limitations 07087 D-Sorbitol All foods: as practically needed. 1. For manufacturing or processing purpose. 2. Not allowed in infant foods. 07088 D-Sorbitol Solution 70% All foods: as practically needed. 1. For manufacturing or processing purpose. 2. Not allowed in infant foods. 07089 D-Xylitol All foods: as practically needed. 1. For manufacturing or processing purpose. 2. Not allowed in infant foods. 07090 D-Mannitol All foods: as practically needed. 1. For manufacturing or processing purpose. 2. Not allowed in infant foods. 07091 Maltitol All foods: as practically needed. 1. For manufacturing or processing purpose. 2. Not allowed in 1 infant foods. 07092 Maltitol Syrup All foods: as practically needed. 1. For (Hydrogenated Glucose Syrup) manufacturing or processing purpose. 2. Not allowed in infant foods. 07093 Isomalt All foods: as practically needed. 1. For (Hydrogenated Palatinose) manufacturing or processing purpose. 2. Not allowed in infant foods. 07094 Lactitol All foods: as practically needed. 1. For manufacturing or processing purpose. 2. Not allowed in infant foods. 07095 Erythritol All foods: as practically needed. 1. For manufacturing or processing purpose. 2. Not allowed in infant foods. 11. Seasoning Agents Code Food Additive Items Scope and Application Standards Limitations 11003 Monosodium L-Aspartate All foods: as practically needed. For manufacturing or processing purpose. 11004 Fumaric Acid All foods: as practically needed.