Protein Structure Prediction
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Biomolecules
CHAPTER 3 Biomolecules 3.1 Carbohydrates In the previous chapter you have learnt about the cell and 3.2 Fatty Acids and its organelles. Each organelle has distinct structure and Lipids therefore performs different function. For example, cell membrane is made up of lipids and proteins. Cell wall is 3.3 Amino Acids made up of carbohydrates. Chromosomes are made up of 3.4 Protein Structure protein and nucleic acid, i.e., DNA and ribosomes are made 3.5 Nucleic Acids up of protein and nucleic acids, i.e., RNA. These ingredients of cellular organelles are also called macromolecules or biomolecules. There are four major types of biomolecules— carbohydrates, proteins, lipids and nucleic acids. Apart from being structural entities of the cell, these biomolecules play important functions in cellular processes. In this chapter you will study the structure and functions of these biomolecules. 3.1 CARBOHYDRATES Carbohydrates are one of the most abundant classes of biomolecules in nature and found widely distributed in all life forms. Chemically, they are aldehyde and ketone derivatives of the polyhydric alcohols. Major role of carbohydrates in living organisms is to function as a primary source of energy. These molecules also serve as energy stores, 2021-22 Chapter 3 Carbohydrade Final 30.018.2018.indd 50 11/14/2019 10:11:16 AM 51 BIOMOLECULES metabolic intermediates, and one of the major components of bacterial and plant cell wall. Also, these are part of DNA and RNA, which you will study later in this chapter. The cell walls of bacteria and plants are made up of polymers of carbohydrates. -

Density Estimation for Protein Conformation Angles Using a Bivariate Von Mises Distribution and Bayesian Nonparametrics
Density Estimation for Protein Conformation Angles Using a Bivariate von Mises Distribution and Bayesian Nonparametrics Kristin P. LENNOX, David B. DAHL, Marina VANNUCCI, and Jerry W. TSAI Interest in predicting protein backbone conformational angles has prompted the development of modeling and inference procedures for bivariate angular distributions. We present a Bayesian approach to density estimation for bivariate angular data that uses a Dirichlet process mixture model and a bivariate von Mises distribution. We derive the necessary full conditional distributions to fit the model, as well as the details for sampling from the posterior predictive distribution. We show how our density estimation method makes it possible to improve current approaches for protein structure prediction by comparing the performance of the so-called ‘‘whole’’ and ‘‘half’’ position dis- tributions. Current methods in the field are based on whole position distributions, as density estimation for the half positions requires techniques, such as ours, that can provide good estimates for small datasets. With our method we are able to demonstrate that half position data provides a better approximation for the distribution of conformational angles at a given sequence position, therefore providing increased efficiency and accuracy in structure prediction. KEY WORDS: Angular data; Density estimation; Dirichlet process mixture model; Torsion angles; von Mises distribution. 1. INTRODUCTION and Ohlson 2002; Lovell et al. 2003; Rother, Sapiro, and Pande 2008), but they behave poorly for these subdivided datasets. Computational structural genomics has emerged as a pow- This is unfortunate, because these subsets provide structure erful tool for better understanding protein structure and func- prediction that is more accurate, as it utilizes more specific tion using the wealth of data from ongoing genome projects. -

POLYMER STRUCTURE and CHARACTERIZATION Professor
POLYMER STRUCTURE AND CHARACTERIZATION Professor John A. Nairn Fall 2007 TABLE OF CONTENTS 1 INTRODUCTION 1 1.1 Definitions of Terms . 2 1.2 Course Goals . 5 2 POLYMER MOLECULAR WEIGHT 7 2.1 Introduction . 7 2.2 Number Average Molecular Weight . 9 2.3 Weight Average Molecular Weight . 10 2.4 Other Average Molecular Weights . 10 2.5 A Distribution of Molecular Weights . 11 2.6 Most Probable Molecular Weight Distribution . 12 3 MOLECULAR CONFORMATIONS 21 3.1 Introduction . 21 3.2 Nomenclature . 23 3.3 Property Calculation . 25 3.4 Freely-Jointed Chain . 27 3.4.1 Freely-Jointed Chain Analysis . 28 3.4.2 Comment on Freely-Jointed Chain . 34 3.5 Equivalent Freely Jointed Chain . 37 3.6 Vector Analysis of Polymer Conformations . 38 3.7 Freely-Rotating Chain . 41 3.8 Hindered Rotating Chain . 43 3.9 More Realistic Analysis . 45 3.10 Theta (Θ) Temperature . 47 3.11 Rotational Isomeric State Model . 48 4 RUBBER ELASTICITY 57 4.1 Introduction . 57 4.2 Historical Observations . 57 4.3 Thermodynamics . 60 4.4 Mechanical Properties . 62 4.5 Making Elastomers . 68 4.5.1 Diene Elastomers . 68 0 4.5.2 Nondiene Elastomers . 69 4.5.3 Thermoplastic Elastomers . 70 5 AMORPHOUS POLYMERS 73 5.1 Introduction . 73 5.2 The Glass Transition . 73 5.3 Free Volume Theory . 73 5.4 Physical Aging . 73 6 SEMICRYSTALLINE POLYMERS 75 6.1 Introduction . 75 6.2 Degree of Crystallization . 75 6.3 Structures . 75 Chapter 1 INTRODUCTION The topic of polymer structure and characterization covers molecular structure of polymer molecules, the arrangement of polymer molecules within a bulk polymer material, and techniques used to give information about structure or properties of polymers. -

Structure Validation Article
14 STRUCTURAL QUALITY ASSURANCE Roman A. Laskowski The experimentally determined three-dimensional (3D) structures of proteins and nucleic acids represent the knowledge base from which so much understanding of biological processes has been derived over the last three decades of the twentieth cen- tury. Individual structures have provided explanations of specific biochemical functions and mechanisms, while comparisons of structures have given insights into general principles governing these complex molecules, the interactions they make, and their biological roles. The 3D structures form the foundation of structural bioinformatics; all structural analyses depend on them and would be impossible without them. Therefore, it is crucial to bear in mind two important truths about these structures, both of which result from the fact that they have been determined experimentally. The first is that the result of any experiment is merely a model that aims to give as good an explanation for the experimental data as possible. The term structure is commonly used, but you should realize that this should be correctly read as model. As such the model may be an accurate and meaningful representation of the molecule, or it may be a poor one. The quality of the data and the care with which the experiment has been performed will determine which it is. Independently performed experiments can arrive at very similar models of the same molecule; this suggests that both are accurate representations, that they are good models. The second important truth is that any experiment, however carefully performed, will have errors associated with it. These errors come in two distinct varieties: sys- tematic and random. -

Protein Sequencing Production of Ions for Mass Spectrometry
Lecture 1 Introduction- Protein Sequencing Production of Ions for Mass Spectrometry Nancy Allbritton, M.D., Ph.D. Department of Physiology & Biophysics 824-9137 (office) [email protected] Office- Rm D349 Medical Science D Bldg. Introduction to Proteins Amino Acid- structural unit of a protein α carbon Amino acids- linked by peptide (amide) bond Amino Acids Proteins- 20 amino acids (Recall DNA- 4 bases) R groups- Varying size, shape, charge, H-bonding capacity, & chemical reactivity Introduction to Proteins Polypeptide Chain (Protein) - Many amino acids linked by peptide bonds By convention: Residue 1 starts at amino terminus. Polypeptides- a. Main chain i.e. regularly repeating portion b. Side chains- variable portion Introduction to Proteins 25,000 human genes >2X106 proteins Natural Proteins - Typically 50-2000 amino acids i.e. 550-220,000 molecular weight Over 200 different types of post-translational modifications. Ex: proteolysis, phosphorylation, acetylation, glycosylation S S S S S S Ex: Insulin Protein Complexity Is Very Large Over 200 different types of post-translational modifications. The Problem of Protein Sequencing. Edman Degradation: Step-wise cleavage of an amino acid from the amino terminus of a peptide. Alanine Gly 1 2 Reacts with uncharged NH3 1 2 Gly 1 2 Cyclizes & Releases in Mild Acid H N-Gly 2 2 2 PTH-Alanine Edman Degradation 1. Must be short peptide (<50 a.a.) amino acid release- 98% efficiency proteins- must fragment (CNBr or trypsin) Trypsin 2. Frequently fails due to a blocked amino terminus 3. Intolerant of impurities 4. Tedious & time consuming (hours-days) 1 amino acid cycle- 2 hours Solution: Mass Spectrometry 1. -

Ideal Chain Conformations and Statistics
ENAS 606 : Polymer Physics Chinedum Osuji 01.24.2013:HO2 Ideal Chain Conformations and Statistics 1 Overview Consideration of the structure of macromolecules starts with a look at the details of chain level chemical details which can impact the conformations adopted by the polymer. In the case of saturated carbon ◦ backbones, while maintaining the desired Ci−1 − Ci − Ci+1 bond angle of 112 , the placement of the final carbon in the triad above can occur at any point along the circumference of a circle, defining a torsion angle '. We can readily recognize the energetic differences as a function this angle, U(') such that there are 3 minima - a deep minimum corresponding the the trans state, for which ' = 0 and energetically equivalent gauche− and gauche+ states at ' = ±120 degrees, as shown in Fig 1. Figure 1: Trans, gauche- and gauche+ configurations, and their energetic sates 1.1 Static Flexibility The static flexibility of the chain in equilibrium is determined by the difference between the levels of the energy minima corresponding the gauche and trans states, ∆. If ∆ < kT , the g+, g− and t states occur with similar probability, and so the chain can change direction and appears as a random coil. If ∆ takes on a larger value, then the t conformations will be enriched, so the chain will be rigid locally, but on larger length scales, the eventual occurrence of g+ and g− conformations imparts a random conformation. Overall, if we ignore details on some length scale smaller than lp, the persistence length, the polymer appears as a continuous flexible chain where 1 lp = l0 exp(∆/kT ) (1) where l0 is something like a monomer length. -

Chapter 13 Protein Structure Learning Objectives
Chapter 13 Protein structure Learning objectives Upon completing this material you should be able to: ■ understand the principles of protein primary, secondary, tertiary, and quaternary structure; ■use the NCBI tool CN3D to view a protein structure; ■use the NCBI tool VAST to align two structures; ■explain the role of PDB including its purpose, contents, and tools; ■explain the role of structure annotation databases such as SCOP and CATH; and ■describe approaches to modeling the three-dimensional structure of proteins. Outline Overview of protein structure Principles of protein structure Protein Data Bank Protein structure prediction Intrinsically disordered proteins Protein structure and disease Overview: protein structure The three-dimensional structure of a protein determines its capacity to function. Christian Anfinsen and others denatured ribonuclease, observed rapid refolding, and demonstrated that the primary amino acid sequence determines its three-dimensional structure. We can study protein structure to understand problems such as the consequence of disease-causing mutations; the properties of ligand-binding sites; and the functions of homologs. Outline Overview of protein structure Principles of protein structure Protein Data Bank Protein structure prediction Intrinsically disordered proteins Protein structure and disease Protein primary and secondary structure Results from three secondary structure programs are shown, with their consensus. h: alpha helix; c: random coil; e: extended strand Protein tertiary and quaternary structure Quarternary structure: the four subunits of hemoglobin are shown (with an α 2β2 composition and one beta globin chain high- lighted) as well as four noncovalently attached heme groups. The peptide bond; phi and psi angles The peptide bond; phi and psi angles in DeepView Protein secondary structure Protein secondary structure is determined by the amino acid side chains. -

Native-Like Mean Structure in the Unfolded Ensemble of Small Proteins
B doi:10.1016/S0022-2836(02)00888-4 available online at http://www.idealibrary.com on w J. Mol. Biol. (2002) 323, 153–164 Native-like Mean Structure in the Unfolded Ensemble of Small Proteins Bojan Zagrovic1, Christopher D. Snow1, Siraj Khaliq2 Michael R. Shirts2 and Vijay S. Pande1,2* 1Biophysics Program The nature of the unfolded state plays a great role in our understanding of Stanford University, Stanford proteins. However, accurately studying the unfolded state with computer CA 94305-5080, USA simulation is difficult, due to its complexity and the great deal of sampling required. Using a supercluster of over 10,000 processors we 2Department of Chemistry have performed close to 800 ms of molecular dynamics simulation in Stanford University, Stanford atomistic detail of the folded and unfolded states of three polypeptides CA 94305-5080, USA from a range of structural classes: the all-alpha villin headpiece molecule, the beta hairpin tryptophan zipper, and a designed alpha-beta zinc finger mimic. A comparison between the folded and the unfolded ensembles reveals that, even though virtually none of the individual members of the unfolded ensemble exhibits native-like features, the mean unfolded structure (averaged over the entire unfolded ensemble) has a native-like geometry. This suggests several novel implications for protein folding and structure prediction as well as new interpretations for experiments which find structure in ensemble-averaged measurements. q 2002 Elsevier Science Ltd. All rights reserved Keywords: mean-structure hypothesis; unfolded state of proteins; *Corresponding author distributed computing; conformational averaging Introduction under folding conditions, with some notable exceptions.14 – 16 This is understandable since under Historically, the unfolded state of proteins has such conditions the unfolded state is an unstable, received significantly less attention than the folded fleeting species making any kind of quantitative state.1 The reasons for this are primarily its struc- experimental measurement very difficult. -
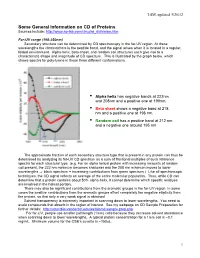
CD of Proteins Sources Include
LSM, updated 3/26/12 Some General Information on CD of Proteins Sources include: http://www.ap-lab.com/circular_dichroism.htm Far-UV range (190-250nm) Secondary structure can be determined by CD spectroscopy in the far-UV region. At these wavelengths the chromophore is the peptide bond, and the signal arises when it is located in a regular, folded environment. Alpha-helix, beta-sheet, and random coil structures each give rise to a characteristic shape and magnitude of CD spectrum. This is illustrated by the graph below, which shows spectra for poly-lysine in these three different conformations. • Alpha helix has negative bands at 222nm and 208nm and a positive one at 190nm. • Beta sheet shows a negative band at 218 nm and a positive one at 196 nm. • Random coil has a positive band at 212 nm and a negative one around 195 nm. The approximate fraction of each secondary structure type that is present in any protein can thus be determined by analyzing its far-UV CD spectrum as a sum of fractional multiples of such reference spectra for each structural type. (e.g. For an alpha helical protein with increasing amounts of random coil present, the 222 nm minimum becomes shallower and the 208 nm minimum moves to lower wavelengths ⇒ black spectrum + increasing contributions from green spectrum.) Like all spectroscopic techniques, the CD signal reflects an average of the entire molecular population. Thus, while CD can determine that a protein contains about 50% alpha-helix, it cannot determine which specific residues are involved in the helical portion. -

9 3 Innovirolgy Examples and Applications
9.3: Examples and applications 1) Now that you have an understanding of the mechanisms behind the phage display system, let’s have a look at how it’s used in research. i) Phage display has a broad spectrum of applications. This can go from improvement of enzymes, to proteome analysis or drug and target discovery but is certainly not restricted to these. 2) The first thing I want to focus on is the optimization of enzymes. A large variety of enzymes are being used today in different fields of industry and biotech. These enzymes are originally made by organisms for a specific function in a specific niche or environment, like inside the cell cytoplasm. Therefore, they often have undesirable properties for the function or environment we want to use them in. They might not work fast enough, be unstable in the conditions they are used in or do not work on the desired substrate. Phage display can aid in improving these enzymes by selecting for example for better thermostability, activity or enhanced substrate binding. i) Let’s start with stability. Take for example an enzyme that is normally present In a cell at cytoplasm at 37°C. However when we use it in an industrial process where we need 50°C, the enzyme becomes unstable and denatures. In phage display we can make a library of mutagenized enzyme and display them on, in this case, the gene 3 protein of the phage. In the binding step of the biopanning most phages will not be able to bind the enzyme substrate at 50°C as they are denatured. -

Single-Molecule Peptide Fingerprinting
Single-molecule peptide fingerprinting Jetty van Ginkela,b, Mike Filiusa,b, Malwina Szczepaniaka,b, Pawel Tulinskia,b, Anne S. Meyera,b,1, and Chirlmin Joo (주철민)a,b,1 aKavli Institute of Nanoscience, Delft University of Technology, 2629HZ Delft, The Netherlands; and bDepartment of Bionanoscience, Delft University of Technology, 2629HZ Delft, The Netherlands Edited by Alan R. Fersht, Gonville and Caius College, Cambridge, United Kingdom, and approved February 21, 2018 (received for review May 1, 2017) Proteomic analyses provide essential information on molecular To obtain ordered determination of fluorescently labeled amino pathways of cellular systems and the state of a living organism. acids, we needed a molecular probe that can scan a peptide in a Mass spectrometry is currently the first choice for proteomic processive manner. We adopted a naturally existing molecular analysis. However, the requirement for a large amount of sample machinery, the AAA+ protease ClpXP from Escherichia coli.The renders a small-scale proteomics study challenging. Here, we ClpXP protein complex is an enzymatic motor that unfolds and demonstrate a proof of concept of single-molecule FRET-based degrades protein substrates. ClpX monomers form a homohexa- + protein fingerprinting. We harnessed the AAA protease ClpXP to meric ring (ClpX6) that can exercise a large mechanical force to scan peptides. By using donor fluorophore-labeled ClpP, we se- unfold proteins using ATP hydrolysis (11, 12). Through iterative quentially read out FRET signals from acceptor-labeled amino acids rounds of force-generating power strokes, ClpX6 translocates of peptides. The repurposed ClpXP exhibits unidirectional process- substrates through the center of its ring in a processive manner ing with high processivity and has the potential to detect low- (13, 14), with extensive promiscuity toward unnatural substrate abundance proteins. -

Intrinsic Indicators for Specimen Degradation
Laboratory Investigation (2013) 93, 242–253 & 2013 USCAP, Inc All rights reserved 0023-6837/13 $32.00 Intrinsic indicators for specimen degradation Jie Li1, Catherine Kil1, Kelly Considine1, Bartosz Smarkucki1, Michael C Stankewich1, Brian Balgley2 and Alexander O Vortmeyer1 Variable degrees of molecular degradation occur in human surgical specimens before clinical examination and severely affect analytical results. We therefore initiated an investigation to identify protein markers for tissue degradation assessment. We exposed 4 cell lines and 64 surgical/autopsy specimens to defined periods of time at room temperature before procurement (experimental cold ischemic time (CIT)-dependent tissue degradation model). Using two-dimen- sional fluorescence difference gel electrophoresis in conjunction with mass spectrometry, we performed comparative proteomic analyses on cells at different CIT exposures and identified proteins with CIT-dependent changes. The results were validated by testing clinical specimens with western blot analysis. We identified 26 proteins that underwent dynamic changes (characterized by continuous quantitative changes, isoelectric changes, and/or proteolytic cleavages) in our degradation model. These changes are strongly associated with the length of CIT. We demonstrate these proteins to represent universal tissue degradation indicators (TDIs) in clinical specimens. We also devised and implemented a unique degradation measure by calculating the quantitative ratio between TDIs’ intact forms and their respective degradation-