Anorectal Malformation (ARM) in Boys
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Anatomy of the Rectum and Anal Canal
BASIC SCIENCE identify the rectosigmoid junction with confidence at operation. The anatomy of the rectum The rectosigmoid junction usually lies approximately 6 cm below the level of the sacral promontory. Approached from the distal and anal canal end, however, as when performing a rigid or flexible sigmoid- oscopy, the rectosigmoid junction is seen to be 14e18 cm from Vishy Mahadevan the anal verge, and 18 cm is usually taken as the measurement for audit purposes. The rectum in the adult measures 10e14 cm in length. Abstract Diseases of the rectum and anal canal, both benign and malignant, Relationship of the peritoneum to the rectum account for a very large part of colorectal surgical practice in the UK. Unlike the transverse colon and sigmoid colon, the rectum lacks This article emphasizes the surgically-relevant aspects of the anatomy a mesentery (Figure 1). The posterior aspect of the rectum is thus of the rectum and anal canal. entirely free of a peritoneal covering. In this respect the rectum resembles the ascending and descending segments of the colon, Keywords Anal cushions; inferior hypogastric plexus; internal and and all of these segments may be therefore be spoken of as external anal sphincters; lymphatic drainage of rectum and anal canal; retroperitoneal. The precise relationship of the peritoneum to the mesorectum; perineum; rectal blood supply rectum is as follows: the upper third of the rectum is covered by peritoneum on its anterior and lateral surfaces; the middle third of the rectum is covered by peritoneum only on its anterior 1 The rectum is the direct continuation of the sigmoid colon and surface while the lower third of the rectum is below the level of commences in front of the body of the third sacral vertebra. -

Mouth Esophagus Stomach Rectum and Anus Large Intestine Small
1 Liver The liver produces bile, which aids in digestion of fats through a dissolving process known as emulsification. In this process, bile secreted into the small intestine 4 combines with large drops of liquid fat to form Healthy tiny molecular-sized spheres. Within these spheres (micelles), pancreatic enzymes can break down fat (triglycerides) into free fatty acids. Pancreas Digestion The pancreas not only regulates blood glucose 2 levels through production of insulin, but it also manufactures enzymes necessary to break complex The digestive system consists of a long tube (alimen- 5 carbohydrates down into simple sugars (sucrases), tary canal) that varies in shape and purpose as it winds proteins into individual amino acids (proteases), and its way through the body from the mouth to the anus fats into free fatty acids (lipase). These enzymes are (see diagram). The size and shape of the digestive tract secreted into the small intestine. varies in each individual (e.g., age, size, gender, and disease state). The upper part of the GI tract includes the mouth, throat (pharynx), esophagus, and stomach. The lower Gallbladder part includes the small intestine, large intestine, The gallbladder stores bile produced in the liver appendix, and rectum. While not part of the alimentary 6 and releases it into the duodenum in varying canal, the liver, pancreas, and gallbladder are all organs concentrations. that are vital to healthy digestion. 3 Small Intestine Mouth Within the small intestine, millions of tiny finger-like When food enters the mouth, chewing breaks it 4 protrusions called villi, which are covered in hair-like down and mixes it with saliva, thus beginning the first 5 protrusions called microvilli, aid in absorption of of many steps in the digestive process. -

Study Guide Medical Terminology by Thea Liza Batan About the Author
Study Guide Medical Terminology By Thea Liza Batan About the Author Thea Liza Batan earned a Master of Science in Nursing Administration in 2007 from Xavier University in Cincinnati, Ohio. She has worked as a staff nurse, nurse instructor, and level department head. She currently works as a simulation coordinator and a free- lance writer specializing in nursing and healthcare. All terms mentioned in this text that are known to be trademarks or service marks have been appropriately capitalized. Use of a term in this text shouldn’t be regarded as affecting the validity of any trademark or service mark. Copyright © 2017 by Penn Foster, Inc. All rights reserved. No part of the material protected by this copyright may be reproduced or utilized in any form or by any means, electronic or mechanical, including photocopying, recording, or by any information storage and retrieval system, without permission in writing from the copyright owner. Requests for permission to make copies of any part of the work should be mailed to Copyright Permissions, Penn Foster, 925 Oak Street, Scranton, Pennsylvania 18515. Printed in the United States of America CONTENTS INSTRUCTIONS 1 READING ASSIGNMENTS 3 LESSON 1: THE FUNDAMENTALS OF MEDICAL TERMINOLOGY 5 LESSON 2: DIAGNOSIS, INTERVENTION, AND HUMAN BODY TERMS 28 LESSON 3: MUSCULOSKELETAL, CIRCULATORY, AND RESPIRATORY SYSTEM TERMS 44 LESSON 4: DIGESTIVE, URINARY, AND REPRODUCTIVE SYSTEM TERMS 69 LESSON 5: INTEGUMENTARY, NERVOUS, AND ENDOCRINE S YSTEM TERMS 96 SELF-CHECK ANSWERS 134 © PENN FOSTER, INC. 2017 MEDICAL TERMINOLOGY PAGE III Contents INSTRUCTIONS INTRODUCTION Welcome to your course on medical terminology. You’re taking this course because you’re most likely interested in pursuing a health and science career, which entails proficiencyincommunicatingwithhealthcareprofessionalssuchasphysicians,nurses, or dentists. -
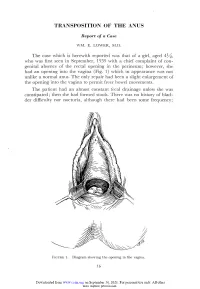
Transposition of the Anus
TRANSPOSITION OF THE ANUS Report of a Case WM. E. LOWER, M.D. The case which is herewith reported was that of a girl, aged 4 who was first seen in September, 1939 with a chief complaint of con- genital absence of the rectal opening in the perineum; however, she had an opening into the vagina (Fig. 1) which in appearance was not unlike a normal anus. The only repair had been a slight enlargement of the opening into the vagina to permit freer bowel movements. The patient had an almost constant fecal drainage unless she was constipated; then she had formed stools. There was no history of blad- der difficulty nor nocturia, although there had been some frequency; 16 Downloaded from www.ccjm.org on September 30, 2021. For personal use only. All other uses require permission. TRANSPOSITION OF THE ANUS and the child had good bladder control. Some voluntary control oi bowel movements also had been observed. In June, 1941 the patient was admitted to the Cleveland Clinic Hospital for operation. Under general anesthesia a loop sigmoid FIGURE 2. Separation of the opening in the vagina and the perineal incision. colostomy was performed, after which the lower bowel was thoroughly cleansed. When the colostomy was functioning well, an opening was made in the perineum, and the opening in the vagina dissected free 17 Downloaded from www.ccjm.org on September 30, 2021. For personal use only. All other uses require permission. WM. E. LOWER FIGURE 3. Freeing the opening into the vagina. (Figs. 2 and 3). With a long forceps this part of the gut was transposed to the new opening in the perineum (Fig. -

Human Body- Digestive System
Previous reading: Human Body Digestive System (Organs, Location and Function) Science, Class-7th, Rishi Valley School Next reading: Cardiovascular system Content Slide #s 1) Overview of human digestive system................................... 3-4 2) Organs of human digestive system....................................... 5-7 3) Mouth, Pharynx and Esophagus.......................................... 10-14 4) Movement of food ................................................................ 15-17 5) The Stomach.......................................................................... 19-21 6) The Small Intestine ............................................................... 22-23 7) The Large Intestine ............................................................... 24-25 8) The Gut Flora ........................................................................ 27 9) Summary of Digestive System............................................... 28 10) Common Digestive Disorders ............................................... 31-34 How to go about this module 1) Have your note book with you. You will be required to guess or answer many questions. Explain your guess with reasoning. You are required to show the work when you return to RV. 2) Move sequentially from 1st slide to last slide. Do it at your pace. 3) Many slides would ask you to sketch the figures. – Draw them neatly in a fresh, unruled page. – Put the title of the page as the slide title. – Read the entire slide and try to understand. – Copy the green shade portions in the note book. 4) -

Congenital Deformities of the Anus and the Rectum*
Arch Dis Child: first published as 10.1136/adc.30.149.42 on 1 February 1955. Downloaded from CONGENITAL DEFORMITIES OF THE ANUS AND THE RECTUM* BY DENIS BROWNE From The Hospital for Sick Children, Great Ormond Street, London One of the regular methods of progression in well recognized one in which fusion is deficient, with medicine is by the analysis of large vaguely assorted the result of a hare-lip or similar deformity. groups of cases into smaller and exactly defined Other groups of deformities, those of the various categories. The process consists in a mixture of imperfect and stenotic anuses, though recognized, observation, abstract reasoning and experiment. are only recognized by few, and are hardly described As instances of this the work of Hamilton Russell at all in textbooks. (1922) may be quoted, when by a combination of The Imperfect Anus observation and abstract reasoning he established the existence of inguinal hernias due to a congenital Stenosis of the Anus. The normal anus of the malformation, and distinguished them from those newborn should take the male adult little finger due to purely mechanical causes, which had been without difficulty, and it may be mentioned that the classed with them. Then there is the work of process of testing this capacity is probably the best Swenson and Bill (1948), which has split up the vague treatment for mild degrees of constipation in theby copyright. group classed under the unhappy name of megacolon small baby. The more severe degrees of stenosis into two classes, one being that of Hirschsprung's are obvious enough if looked for, though they may disease, a congenital deformity consisting in the escape this investigation for months, with the grave absence of ganglion cells in the bowel, and the other danger of producing the obstinate condition of a functional failure to empty a normal bowel which colonic inertia through loss of the normal irritability can be called 'colonic inertia'. -

Anatomy of Anal Canal
Anatomy of Anal Canal Dr Garima Sehgal Associate Professor Department of Anatomy King George’s Medical University, UP, Lucknow DISCLAIMER: • The presentation includes images which are either hand drawn or have been taken from google images or books. • They are being used in the presentation only for educational purpose. • The author of the presentation claims no personal ownership over images taken from books or google images. • However, the hand drawn images are the creation of the author of the presentation Subdivisions of the perineum • Transverse line joining the anterior part of ischial tuberosities divides perineum into: 1. Urogenital region / triangle- ANTERIORLY 2. Anal region / triangle - POSTERIORLY Anal canal may be affected by many conditions that are not so rare, not necessarily serious and endangering to life but on the contrary very INCAPACITATING Haemorrhoids Anal fistula Anal fissure Perianal abscess Learning objectives At the end of this teaching session on anatomy of Anal canal all the MBBS 1st Year students must be able to correctly: • Describe the location, extent and dimensions of the anal canal • Enumerate the relations of the anal canal • Enumerate the subdivisions of anal canal • Describe & Diagrammatically display the special features on the interior of the anal canal • Discuss the importance of pectinate / dentate line • Write a short note on the arterial supply, venous drainage, nerve supply & lymphatic drainage • Write a short note on the sphincters of the anal canal • Describe the anatomical basis of internal -

Normal Gastrointestinal Motility and Function Esophagus
Normal Gastrointestinal Motility and Function "Motility" is an unfamiliar word to many people; it is used primarily to describe the contraction of the muscles in the gastrointestinal tract. Because the gastrointestinal tract is a circular tube, when these muscles contract, they close off the tube or make the opening inside smaller - they squeeze. These muscles can contract in a synchronized way to move the food in one direction (usually downstream, but occasionally upstream for short distances); this is called peristalsis. If you looked at the intestine, you would see a ring of contraction that moves along pushing contents ahead of it. At other times, the muscles in adjacent parts of the gastrointestinal tract squeeze more or less independently of each other: this has the effect of mixing the contents but not moving them up or down. Both kinds of contraction patterns are called motility. The gastrointestinal tract is divided into four distinct parts: the esophagus, stomach, small intestine, and large intestine (colon). They are separated from each other by special muscles called sphincters which normally stay tightly closed and which regulate the movement of food and food residues from one part to another. Each part of the gastrointestinal tract has a unique function to perform in digestion, and as a result each part has a distinct type of motility and sensation. When motility or sensations are not appropriate for performing this function, they cause symptoms such as bloating, vomiting, constipation, or diarrhea which are associated with subjective sensations such as pain, bloating, fullness, and urgency to have a bowel movement. -

The Digestive System
THE DIGESTIVE SYSTEM COMPILED BY HOWIE BAUM DIGESTIVE SYSTEM People are probably more aware of their digestive system than of any other system, not least because of its frequent messages. Hunger, thirst, appetite, gas ☺, and the frequency and nature of bowel movements, are all issues affecting daily life. The Digestive Tract • Six Functions of the Digestive System 1. Ingestion 2. Mechanical processing 3. Digestion 4. Secretion 5. Absorption 6. Excretion The Digestive Tract • Ingestion – Occurs when materials enter digestive tract via the mouth • Mechanical Processing – Crushing and shearing – Makes materials easier to propel along digestive tract • Digestion – The chemical breakdown of food into small organic fragments for absorption by digestive epithelium The Digestive Tract • Secretion – Is the release of water, acids, enzymes, buffers, and salts – By epithelium of digestive tract – By glandular organs • Absorption – Movement of organic substrates, electrolytes, vitamins, and water – Across digestive epithelium tissue – Into the interstitial fluid of digestive tract • Excretion – Removal of waste products from body fluids – Process called defecation removes feces AN INTRODUCTION TO THE DIGESTIVE SYSTEM • The Digestive Tract • Also called the gastrointestinal (GI) tract or alimentary canal • Is a muscular tube • Extends from our mouth to the anus • Passes through the pharynx, esophagus, stomach, and small and large intestines The digestive system is one of the most clearly defined in the body. It consists of a long passageway, the digestive -

Urinary Retention
Urinary Retention National Kidney and Urologic Diseases Information Clearinghouse What is urinary retention? What is the urinary tract Urinary retention is the inability to and how does it work? empty the bladder completely. Urinary The urinary tract is the body’s drainage retention can be acute or chronic. Acute system for removing urine, which is urinary retention happens suddenly and composed of wastes and extra fluid. In lasts only a short time. People with acute order for normal urination to occur, all urinary retention cannot urinate at all, body parts in the urinary tract need to work even though they have a full bladder. together in the correct order. Acute urinary retention, a potentially life-threatening medical condition, Kidneys. The kidneys are two bean-shaped requires immediate emergency treatment. organs, each about the size of a fist. They Acute urinary retention can cause great are located just below the rib cage, one discomfort or pain. on each side of the spine. Every day, the kidneys filter about 120 to 150 quarts of Chronic urinary retention can be a long- blood to produce about 1 to 2 quarts of lasting medical condition. People with urine. The kidneys work around the clock; chronic urinary retention can urinate. a person does not control what they do. However, they do not completely empty all of the urine from their bladders. Ureters. Ureters are the thin tubes of Often people are not even aware they muscle—one on each side of the bladder— have this condition until they develop that carry urine from each of the kidneys to another problem, such as urinary the bladder. -

Anathomy and Phisiology of Digestive System
Danijel Borković,dr.med. Prague, 10.october 2016 Anathomy and phisiology of digestive system Digestive system: a) series of hollow organs joined in a long, twisting tube (gastointestinal tract) - food passes through them b) accessory organs – food doesnt pass through them • Other „helpers“: nerves, hormones, blood, bacteria in GI tract • Digestive system turns food and drink into nutrients (carbohydrates, protein, fats and vitamins) which body uses for energy, cell repair and growth. Anathomy and phisiology of digestive system Gastrointestinal tract: 1.Upper GI tract: a) Mouth b) Throat (pharynx) c) Esophagus d) Stomach 2. Lower GI tract: a) Small intestine b) Large intestine (with rectum) c) Anus Anathomy and phisiology of digestive system Six major functions take place in digestive system: a) Ingestion of food b) Secretion of fluids and digestyve enzymes c) Mixing and movement of food and wastes through the body d) Digestion of food into smaller pieces e) Absorption of nutrients f) Excretion of wastes Anathomy and phisiology of digestive system Mouth (oral cavity): a) Teeth b) The tongue c) Salivary glands Function: a) Chewing food: breaks it into pieces which are more easily digested. b) Saliva: mixes food to begin process of breaking it down in a form our body can absorb and use. Anathomy and phisiology of digestive system • Throat (pharynx): epiglottis as a switch between GI and RI tract • Esophagus: muscular tube extending from the pharynx to stomach. • Function: a) Deliveres food to stomach with series of contractions – peristalsis. -

Colon and Rectum, Esophagus, Stomach, Anus, Pancreas
SEER Program Coding and Staging Manual 2021 Site-Specific Codes for Neoadjuvant Therapy Treatment Effect Schemas: Colon and Rectum, Esophagus, Stomach, Anus, Pancreas Neoadjuvant Therapy--Treatment Effect data item [NAACCR # 1634] is related to the Neoadjuvant Therapy data item [NAACCR # 1632]. This data item records the findings from the post neoadjuvant therapy surgical pathology report ONLY when surgery is performed after neoadjuvant therapy. This set of codes applies to the schemas: Colon and Rectum, Esophagus, Stomach, Anus, and Pancreas. Code Description 0 Post neoadjuvant surgery not performed 1 Present: No viable cancer cells Complete response Score 0 2 Present: Single cells or rare small groups of cancer cells Near complete response Score 1 3 Present: Residual cancer with evident tumor regression, but more than single cells or rare small groups of cancer cells Partial response Minimal response Score 2 4 Absent: Extensive residual cancer with no evident tumor regression Poor or no response Score 3 6 Neoadjuvant therapy completed and surgical resection performed, response not documented or unknown Cannot be determined 7 Neoadjuvant therapy completed and planned surgical resection not performed 9 Unknown if neoadjuvant therapy performed Unknown if planned surgical procedure performed after completion of neoadjuvant therapy Death Certificate only (DCO) For purposes of this data item, neoadjuvant therapy is defined as systemic treatment (chemotherapy, endocrine/hormone therapy, targeted therapy, immunotherapy, or biological therapy) and/or radiation therapy given to shrink a tumor before surgical resection. Surgical resection: For purposes of this data item, surgical resection is defined as the most definitive surgical procedure that removes some or all of the primary tumor or site, with or without lymph nodes and/or distant metastasis.