Genome-Wide Methylation Profiling Identified Novel Differentially
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Climate Research 57:123
Vol. 57: 123–132, 2013 CLIMATE RESEARCH Published August 20 doi: 10.3354/cr01174 Clim Res Extreme sea ice events in the Chinese marginal seas during the past 2000 years Jie Fei1,2,*, Zhong-Ping Lai2,3, David D. Zhang4, Hong-Ming He5 1Institute of Chinese Historical Geography, Fudan University, Shanghai 200433, PR China 2State Key Laboratory of Cryospheric Sciences (CAREERI), Chinese Academy of Sciences, Lanzhou 730000, PR China 3Qinghai Institute of Salt Lakes, Chinese Academy of Sciences, Xining 810008, PR China 4Department of Geography, The University of Hong Kong, Hong Kong SAR 5Institute of Soil and Water Conservation, Chinese Academy of Sciences & Ministry of Water Resources, Yangling District 712100, PR China ABSTRACT: We used Chinese historical literature to examine extreme records of sea ice events in the Chinese marginal seas during the past 2000 yr. We identified a total of 6 sea ice events that occurred in the sea areas to the south of 35° N. These extreme events occurred in the winters of AD 821/822, 903/904, 1453/1454, 1493/1494, 1654/1655 and 1670/1671. According to the histori- cal records, the southern limit of sea ice in the Chinese marginal seas should be in Hangzhou Bay (30−31° N), and most probably near Haiyan County in Zhejiang Province (30.5° N). The sea ice events of 1453/1454 and 1654/1655 were synchronous with the freezing events of Taihu Lake, and the sea ice event of 1670/1671 was synchronous with the freezing event of the lower reaches of the Yangtze River. However, none of the sea ice events was synchronous with the abnormally early freezing dates of Suwa Lake in Japan or the extreme freezing events of Venice Lagoon in Italy. -

TIER2 SITE NAME ADDRESS PROCESS M Ns Garments Printing & Embroidery
TIER 2 MANUFACTURING SITES - Produced July 2021 TIER2 SITE NAME ADDRESS PROCESS Bangladesh Mns Garments Printing & Embroidery (Unit 2) House 305 Road 34 Hazirpukur Choydana National University Gazipur Manufacturer/Processor (A&E) American & Efird (Bd) Ltd Plot 659 & 660 93 Islampur Gazipur Manufacturer/Processor A G Dresses Ltd Ag Tower Plot 09 Block C Tongi Industrial Area Himardighi Gazipur Next Branded Component Abanti Colour Tex Ltd Plot S A 646 Shashongaon Enayetnagar Fatullah Narayanganj Manufacturer/Processor Aboni Knitwear Ltd Plot 169 171 Tetulzhora Hemayetpur Savar Dhaka 1340 Manufacturer/Processor Afrah Washing Industries Ltd Maizpara Taxi Track Area Pan - 4 Patenga Chottogram Manufacturer/Processor AKM Knit Wear Limited Holding No 14 Gedda Cornopara Ulail Savar Dhaka Next Branded Component Aleya Embroidery & Aleya Design Hose 40 Plot 808 Iqbal Bhaban Dhour Nishat Nagar Turag Dhaka 1230 Manufacturer/Processor Alim Knit (Bd) Ltd Nayapara Kashimpur Gazipur 1750 Manufacturer/Processor Aman Fashions & Designs Ltd Nalam Mirzanagar Asulia Savar Manufacturer/Processor Aman Graphics & Design Ltd Nazimnagar Hemayetpur Savar Dhaka Manufacturer/Processor Aman Sweaters Ltd Rajaghat Road Rajfulbaria Savar Dhaka Manufacturer/Processor Aman Winter Wears Ltd Singair Road Hemayetpur Savar Dhaka Manufacturer/Processor Amann Bd Plot No Rs 2497-98 Tapirbari Tengra Mawna Shreepur Gazipur Next Branded Component Amantex Limited Boiragirchala Sreepur Gazipur Manufacturer/Processor Ananta Apparels Ltd - Adamjee Epz Plot 246 - 249 Adamjee Epz Narayanganj -
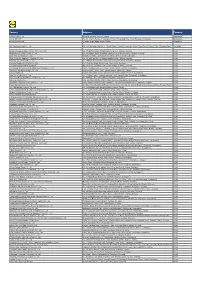
Factory Address Country
Factory Address Country Durable Plastic Ltd. Mulgaon, Kaligonj, Gazipur, Dhaka Bangladesh Lhotse (BD) Ltd. Plot No. 60&61, Sector -3, Karnaphuli Export Processing Zone, North Potenga, Chittagong Bangladesh Bengal Plastics Ltd. Yearpur, Zirabo Bazar, Savar, Dhaka Bangladesh ASF Sporting Goods Co., Ltd. Km 38.5, National Road No. 3, Thlork Village, Chonrok Commune, Korng Pisey District, Konrrg Pisey, Kampong Speu Cambodia Ningbo Zhongyuan Alljoy Fishing Tackle Co., Ltd. No. 416 Binhai Road, Hangzhou Bay New Zone, Ningbo, Zhejiang China Ningbo Energy Power Tools Co., Ltd. No. 50 Dongbei Road, Dongqiao Industrial Zone, Haishu District, Ningbo, Zhejiang China Junhe Pumps Holding Co., Ltd. Wanzhong Villiage, Jishigang Town, Haishu District, Ningbo, Zhejiang China Skybest Electric Appliance (Suzhou) Co., Ltd. No. 18 Hua Hong Street, Suzhou Industrial Park, Suzhou, Jiangsu China Zhejiang Safun Industrial Co., Ltd. No. 7 Mingyuannan Road, Economic Development Zone, Yongkang, Zhejiang China Zhejiang Dingxin Arts&Crafts Co., Ltd. No. 21 Linxian Road, Baishuiyang Town, Linhai, Zhejiang China Zhejiang Natural Outdoor Goods Inc. Xiacao Village, Pingqiao Town, Tiantai County, Taizhou, Zhejiang China Guangdong Xinbao Electrical Appliances Holdings Co., Ltd. South Zhenghe Road, Leliu Town, Shunde District, Foshan, Guangdong China Yangzhou Juli Sports Articles Co., Ltd. Fudong Village, Xiaoji Town, Jiangdu District, Yangzhou, Jiangsu China Eyarn Lighting Ltd. Yaying Gang, Shixi Village, Shishan Town, Nanhai District, Foshan, Guangdong China Lipan Gift & Lighting Co., Ltd. No. 2 Guliao Road 3, Science Industrial Zone, Tangxia Town, Dongguan, Guangdong China Zhan Jiang Kang Nian Rubber Product Co., Ltd. No. 85 Middle Shen Chuan Road, Zhanjiang, Guangdong China Ansen Electronics Co. Ning Tau Administrative District, Qiao Tau Zhen, Dongguan, Guangdong China Changshu Tongrun Auto Accessory Co., Ltd. -

459824 1 En Bookbackmatter 287..294
Postscript The research and writing relating to the major issue “The Chinese Dream and Zhejiang’s Practice” (Society Volume), jointly conducted by the Party Committee of Zhejiang Province and the Chinese Academy of Social Sciences, has lasted for more than half a year since April when it was officially initiated. In the Research Group, Chen Guangjin, the Director of the Institute of Sociology of the Chinese Academy of Social Sciences, serves as the leader, and Yang Jianhua, the Director and research fellow of the Institute of Public Policy of the Zhejiang Academy of Social Sciences, a member of the Advisory Committee of the Zhejiang Provincial People’s Government, serves as the deputy leader. Under the careful guidance of the leaders from the Chinese Academy of Social Sciences, the Party Committee of Zhejiang Province, the Department of Publicity under the Party Committee of Zhejiang Province and the Zhejiang Academy of Social Sciences, with great efforts of all the members of the group, the Research Group ultimately finished the task of research and writing on schedule. In the research project, we resolutely followed the overall requirements of the leaders at various levels, especially the specific requirements raised by Hu Jian, the Executive Deputy Director of the Department of Publicity under the Party Committee of Zhejiang Province, about the research and writing relating to the Society Volume; we earnestly learnt the thoughts and theories concerning social development developed by General Secretary Xi Jinping and his two works Carrying out Solid Work to Stay Ahead, Zhijiang Xinyu, summarized the thoughts, experiments and the practical experience of comrade Xi Jinping, the practical experience of the subsequent successive Party Committees of Zhejiang Province in inheritance and carrying through one blueprint, and presented the great significance of these practices and experience for Zhejiang’s modernization, inspirations and significance for reference for national development. -

Company Brochure-CCPIT Zhejiag 1105
Contents Ⅰ Textile and Garment 1. Stage Group Co., Ltd. 2. Wuchan Zhongda E-commerce Co.,Ltd 3. Zhejiang Shenzhou Woollen Textile Co., Ltd. 4. Zhejiang Linglong Textile Co., Ltd. 5. Zhejiang Tongxing Textile Science & Technology Development Co., Ltd. 6. Kaidi Silk Industry Co., LTD. 7. Mingda Group Co., Ltd. 8. Shaoxing Bolihao Hometextiles Co.,Ltd. 9. Jinhua Shijun Industrial Co., Ltd. 10. Jinhua Aishang Leather Goods Co.,Ltd 11. Pujiang Rongsheng Garment Factory Ⅱ Infrastructure and Energy 12. Hangzhou Dongheng Industrial Group Co., Ltd. 13. ZhouShan Marine Comprehensive Development and Investment Co.,Ltd. 14. Zhejiang Deye Investment Co., Ltd. 15. Zhoushan Water Group Co., Ltd.( cancel) 16. Jiashan Huayang Wooden&flocking Co.,Ltd. Ⅲ Processing and Manufacturing 17. Xinmingli Lighting Co.,Ltd. 18. Zhejiang Pan Casa Co.,Ltd. 19. Zhejiang L'meri Home Co.,Ltd. 20. Jinhua Ailanjie Automation Equipment Technology Co., Ltd. 21. Yongkang Arda Motor Co.,Ltd. 22. Zhejiang Myhome Kitchen Co.,Ltd. 23. Quzhou T-nine Tools Co.,Ltd. 24. Zhejiang Chengyuan Heavy Machinery Joint-Stock Co., Ltd. ⅣTrade andLogistics& Cultural and Tourism 25. Zhejiang Everbirght Development Corp. 26. Zhejiang Kaihom Logistics Co.,Ltd. 27. Zhejiang Runjiu Shipping Co., Ltd. 28. lulu&bewell 29. Jinhua Qun'an Trading Co.,Ltd. 30. Zhejiang Pingshanjinshui Culture& Communication Broadcast Co.,Ltd. ⅤElectronic Information and Security Services 31. Zhejiang Chinajoiner Information Technology Co., Ltd. 32. Zhejang Sunhope Electronic Technology Co., Ltd. 33. POPP Electric Co., Ltd.(cancel) 34. Zhejiang XINAN Intelligent Technology Co., Ltd. 35. Jinhua Jindong Security Service Co., Ltd. ⅥChemical Engineering and Medical Equipment 36. Zhejiang Polyker Fluorine Material Co., Ltd. -

September 2018 Factory Name Address Country Department Worker Category Dophil Xafzotaj, Rruga Shjak, Durress Albania Footwear Le
September 2018 Factory Name Address Country Department Worker Category Dophil Xafzotaj, Rruga Shjak, Durress Albania Footwear Less than 1000 workers Ama Syntex Ltd. Plot No. 936 to 939, Vill: Jarun, Kashimpur, Joydebpur, Gazipur, Dhaka Bangladesh Apparel Less than 1000 workers A-One (BD) Limited Plot No. 114-120, Depz, Ganakbari, Savar, Dhaka Bangladesh Apparel 1001- 5000 workers Apparel Plus Ltd. (A T S APPARELS LIMITED) Holding No. C-18 Dilan Complex, Chandona Chowrasta, Dhaka Road, Gazipur Bangladesh Apparel 1001- 5000 workers BD Designs Private Limited Plot No. 48-49, Sector-3, Karnaphuli Epz, Chittagong Bangladesh Apparel 1001- 5000 workers Continental Garments Ind (Pvt.) Ltd. 8, Dewan Idris Road, Boro Rangamatia, Ashulia, Savar, Dhaka Bangladesh Apparel 1001- 5000 workers Creative Woolwear 3/B Darus Salam Road, Mirpur-1 Dhaka Bangladesh Apparel Less than 1000 workers Dird Composite Textiles Ltd. Shatiabari, Dhaladia, Rajendrapur, Sreepur, Gazipur Bangladesh Apparel 1001- 5000 workers DNV Clothing Ltd. Plot No. 100-101, Adamjee EPZ, Siddhirgonj, Narayangonj Bangladesh Apparel 1001- 5000 workers Echotex Ltd. Chandra, Palli Biddut, Kaliakoir, Gazipur, Dhaka Bangladesh Apparel 5001- 10,000 workers Fakir Apparels Ltd. A-142-145, B-501-503, BSCIC Industrial Estate, Fatullah, Narayanganj Bangladesh Apparel 5001-10,000 workers Far East Knitting & Dyeing Industries Ltd. Polli Bidut, Purbo Chandra, Shafipur, Kaliakoir, Gazipur Bangladesh Apparel 5001- 10,000 workers Fortis Garments Ltd. Holiding No. 100/1, Block B, Saheed Mosarraf Hossain Road, Purbo Chandora, Sofipur, Kaliakoir, Gazipur Bangladesh Apparel 1001- 5000 workers Global Outerwear Ltd. 89, Berulia Road, Savar, Dhaka Bangladesh Apparel 1001- 5000 workers Golden Times Sweater & Dyeing Ltd. Mulaied, Mawna, Chowrasta, Sreepur, Gazipur, Dhaka Bangladesh Apparel Less than 1000 workers Hams Garments Ltd. -

FACTORY LIST Tier 1 Trade Partners
FACTORY LIST Tier 1 Trade Partners Product Parent Company Facility Name Facility Address Province Country Number of Category Workers Apparel Calzificio Telemaco SRL Calzificio Telemaco SRL Via Brentella 9, Cap 31040, Trevignano Treviso Italy < 50 Accessories B.S.T. International Trade Company Ltd Chanchang Stainless Steel Products Co., Ltd Xiangzhu Industrial Zone, Yongkang City 322300 Zhejiang China < 50 Apparel Changsu Gloves Factory Co. Changsu Gloves Factory Co. No. 199, Xichun Lane, Hupu Lane, Bixi New District, Changshu City 2115500 Jiangsu China < 50 Apparel Changsu Haozu Socks Co., Ltd Changsu Haozu Socks Co., Ltd No. 6, Minda Road, XushiQigan Village, Dongbin Town, Changshu City 2115500 Jiangsu China < 50 Apparel Castlecrafts Ltd T/A Dewhirst Menswear Dalian Taiyangying Garments Co.,Ltd Chengguan Community, Pikou Town Sub-district, Pulandian District, Dalian Liaoning China 200 - 300 Accessories Delta (Tien Giang) Co., Ltd Delta (Tien Giang) Co., Ltd 69B&70A, Long jiang Industrial Park, Tan Lap 1 valley, Tan Phuoc County, Tiền Giang Vietnam 100 - 200 Accessories Favour Light Co.,Ltd DongGuan Sheng Shi Hardware & Plastics Prod. Co. Ltd No.3 Daling Street, Jiao Yi Tang Administrative District, Tang Xia Town Guangdong China 800 - 900 Accessories Dongguan Yifeng Lock Co. Ltd Dongguan Yifeng Lock Co. Ltd Building A-H, No.28, Street 6, First Gongye Road, Tutang Village, Changping Town, Dongguan Guangdong China 1100 - 1200 Apparel DongYang New DongHuang Garment Co., Ltd DongYang New DongHuang Garment Co., Ltd No.52 Jinwang Road, West -
Bestseller Factory List
BESTSELLER FACTORY LIST LAST UPDATED: 21.05.2021 It is imperative for us to work with our partners in an open and honest way. We continuously seek to create more transparency in our supply chain to address risks and promote positive change. In light of this, and to provide increased transparency, we are making supplier factory information publicly available. The factory list includes the name, address, product type and number of workers of all tier 1 manufacturing factories (cut-make-trim) of apparel, footwear and accessories. The list will be updated twice a year. As changes occur in our supply chain with new partners, new factories onboarded, or factories being phased out, such alterations will be collected and published in the next supplier factory list update. FACTORY NAME ADDRESS MALE FEMALE NO. OF POSTAL CITY REGION COUNTRY PRODUCT EMPLOYEE EMPLOYEE EMPLOYEES CODE TYPES % % Almeg Shoes No. 14 Rruga Albania 5 95 <500 2000 Durres Durres ALBANIA Footwear Aba Fashions Ltd. No. 521/1 Gacha, Gazipur City Corporation 35 65 1001-5000 1704 Gazipur Dhaka BANGLADESH Apparel ABM Fashions Ltd. No.1143 &1145 Kashimpur Road, Konabari, Gazipur 30 70 1001-5000 1700 Dhaka Dhaka BANGLADESH Apparel Agami Apparels Ltd. Nayapara, Kathgara, Ashulia 40 60 5001-10000 1344 Savar Dhaka BANGLADESH Apparel Agami Fashions Ltd. Abdus Sattar Road, Pallibiduyt, Holding No. 79/1 30 70 1001-5000 1751 Gazipur Dhaka BANGLADESH Apparel AKM Knit Wear Ltd. No. 14 Gedda Karnapara, Ulail, Savar 35 65 >10000 1340 Dhaka Dhaka BANGLADESH Apparel Al-Muslim Apparels Ltd. No. 12 Nischintapur 25 75 <500 1341 Ashulia Dhaka BANGLADESH Apparel Aman Tex Ltd. -

Indirect China Suppliers
INDIRECT CHINA SUPPLIERS Here below you can find upstream suppliers that are located in China and operations like wet pro- cessing, wastewater treatment and hazardous waste treatment. The list includes suppliers that have been contracted by our direct suppliers and who perform operations that our main suppliers are not capable of doing in their own facilities. SUPPLIER COUNTRY SUPPLIER TYPE Changzhou Dawei Environmental Technology Co. Jiangsu, China Hazardous Waste Plant Cervo Textile Jiangsu, China Fabric Mill Dalian Dongtai Industrial Waste Treatment Co., Ltd. Liaoning, China Hazardous Waste Plant Dystar Wuxi Colours Co.,Ltd. Jiangsu, China Dye Supplier Hangzhou Zhongao Yasheng Sweater Co Ltd Zhejiang, China Fabric Mill Jiashan County Dadi Wushui Treatment Project Co., Ltd. Zhejiang, China Waste Water Treatment Plant Jiaxing Xinjia Aisi Thermoelectricity Co., Ltd. Zhejiang, China Hazardous Waste Plant Jiangsu Longda Transfer Printing Textile Co.,Ltd. Jiangsu, China Fabric Mill Jiangsu Yueda Cotton Spinning Co.,Ltd. Jiangsu, China Yarn Supplier Jiangyin Jintong Petroleum Chemical Co., Ltd. Jiangsu, China Hazardous Waste Plant LUTHAI TEXTILE CO.,LTD. Shandong, China Fabric Mill Ningbo Huake Textile Auxiliary Co Ltd Zhejiang, China Dye Supplier Yiyang Binba Textile Co., Ltd. Zhejiang, China Lining Maker Zhejiang Rihua Chemistry Co.,Ltd. Zhejiang, China Dye Supplier Poly Beauty Textile Ltd. Guangdong, China Lining Maker Qihe New Materials Technology Zhejiang, China Dye Supplier Shengzhou Drainage Management Co., Ltd. Zhejiang, China Waste Water Treatment Plant WUXI HUILING CASHMERE ADORNMENT CO., LTD Jiangsu, China Fabric Mill Wujiang Haosheng Jet Weaving Co., Ltd. Jiangsu, China Fabric Mill ZHEJIANG BABEI SILK INDUSTRY CO.,LTD Zhejiang, China Yarn Supplier Zhejiang Houyuan Textiles Inc. -

Vertical Facility List
Facility List The Walt Disney Company is committed to fostering safe, inclusive and respectful workplaces wherever Disney-branded products are manufactured. Numerous measures in support of this commitment are in place, including increased transparency. To that end, we have published this list of the roughly 7,600 facilities in over 70 countries that manufacture Disney-branded products sold, distributed or used in our own retail businesses such as The Disney Stores and Theme Parks, as well as those used in our internal operations. Our goal in releasing this information is to foster collaboration with industry peers, governments, non- governmental organizations and others interested in improving working conditions. Under our International Labor Standards (ILS) Program, facilities that manufacture products or components incorporating Disney intellectual properties must be declared to Disney and receive prior authorization to manufacture. The list below includes the names and addresses of facilities disclosed to us by vendors under the requirements of Disney’s ILS Program for our vertical business, which includes our own retail businesses and internal operations. The list does not include the facilities used only by licensees of The Walt Disney Company or its affiliates that source, manufacture and sell consumer products by and through independent entities. Disney’s vertical business comprises a wide range of product categories including apparel, toys, electronics, food, home goods, personal care, books and others. As a result, the number of facilities involved in the production of Disney-branded products may be larger than for companies that operate in only one or a limited number of product categories. In addition, because we require vendors to disclose any facility where Disney intellectual property is present as part of the manufacturing process, the list includes facilities that may extend beyond finished goods manufacturers or final assembly locations. -

Factory Name Company Name Address AKH ECO APPARELS LTD 495, BALITHA, SHAH BELISHWER, DHAMRAI, DHAKA-1800 DHAKA BANGLADESH AMAN
Factory Name BANGLADESH Company Name Address AKH ECO APPARELS LTD 495, BALITHA, SHAH BELISHWER, DHAMRAI, DHAKA-1800 DHAKA BANGLADESH AMAN GRAPHICS & DESIGNS LTD NAZIMNAGAR HEMAYETPUR SAVAR DHAKA 1340BANGLADESH AMAN KNITTINGS LTD KULASHUR, HEMAYETPUR SAVAR DHAKA BANGLADESH ARRIVAL FASHION LTD BUILDING 1, KOLOMESSOR, BOARD BAZAR GAZIPUR DHAKA 1704BANGLADESH BHIS APPARELS LTD 671, DATTA PARA, HOSSAIN MARKET TONGI GAZIPUR 1712BANGLADESH CORTZ APPARELS LTD (1) - NAWJOR NAWJOR, KADDA BAZAR GAZIPUR BANGLADESH ENERGYPAC FASHIONS LTD BOKRAN MONIPUR, HOTAPARA, GAZIPUR SADAR, GAZIPUR. 1749BANGLADESH ETTADE JEANS LTD 1670/2091, BUILDING NUMBER 3, WEST SHOLASHAHAR, ATURAR DEPOT CHITTAGONG CHITTAGONG 4211BANGLADESH FLAXEN DRESS MAKER LTD AKBOR TOWER, 51-52 SHAH KABIR MAZAR ROAD MOUSHAIR, DAKKHINKHAN DHAKA 1230BANGLADESH HELA CLOTHING BANGLADESH SECTOR:1, PLOT: 53,54,66,67 CHITTAGONG BANGLADESH INDOCHINE TEXTILES (BD) LTD, ASIA EXCEL TRADING LTD PLOT 16-18, DAKHIN PANISHAIL, EPZ-KALIAKAIR ROAD KASHIMPUR GAZIPUR 1349BANGLADESH KDS FASHION LTD 253 / 254, NASIRABAD I/A, AMIN JUTE MILLS, BAYEZID, CHITTAGONG CHITTAGONG CHITTAGONG 4211BANGLADESH LIDA TEXTILE & DYEING LTD HOLDING # 1 (3RD & 4TH FLOOR), BLOCK - C, PURBO ,CHANDONA, SHAFIPUR, KALIAKOIR, DHAKA 1206BANGLADESH MAJUMDER GARMENTS LTD. 113/1, MUDAFA PASCHIM PARA, TONGI. GAZIPUR. 1711BANGLADESH MILLENNIUM TEXTILES (SOUTHERN) LTD BARA RANGAMATIA, ZIRABO, ASHULIA SAVAR DHAKA 1341BANGLADESH NAFA APPARELS LTD-2 VILLAGE-JOYPURA, UNION-SHOMBAG, UPAZILA-DHAMRAI, DISTRICT DHAKA BANGLADESH NOMAN TERRY TOWEL MILLS LTD VAWAL MIRZAPUR GAZIPUR 1703BANGLADESH PANDORA SWEATERS LTD GILAR CHALA, MASTER BARI, SHREEPUR GAZIPUR 1740BANGLADESH PRETTY SWEATERS LTD KULIAR CHAR TOWER, CHAYDANA,GAZIPUR-1704 GAZIPUR 1704BANGLADESH RENAISSANCE APPARELS LTD C-S PLOT #659 & 660, MOUZA-93, RI PARK , ISLAMPUR GAZIPUR SADAR GAZIPUR 1700BANGLADESH RENAISSANCE APPARELS LTD DEWAN EDRIS ROAD, ZIRABO ASHULIA, SAVER DHAKA 1341BANGLADESH ROVERCO (HK) LTD TALTOLI, MONIPUR, B.K.BARI, MIRZAPUR, GAZIPUR 1710. -

Annual Report
封面圖為浙江舟山跨海大橋,全長 46公里並於2010年2月6日通車, 連接寧波和舟山及中間的島嶼。 2020 年,舟山跨海大橋平均每日全程車流量為 XX 輛。 REPORT ANNUAL Definition of Terms 2 Company Profile 4 Corporate Structure of the Group 5 Review of Major Corporate Events 6 Particulars of Major Road Projects 8 Financial and Operating Highlights 10 Chairman’s Statement 13 Management Discussion and Analysis 17 Principal Risks and Uncertainties 38 Corporate Governance Report 42 Directors, Supervisors and Senior Management Profiles 58 Report of the Directors 74 Report of the Supervisory Committee 84 Connected Transactions 86 Independent Auditor’s Report 101 Consolidated Financial Statements & Notes 108 Independent Auditor’s Report 264 (Issued by a third country auditor registered with the UK Financial Reporting Council) Corporate Information 271 Location Map of Expressways in Zhejiang Province 273 Definition of Terms Associate has the meaning ascribed to it under the Listing Rules Audit Committee the audit committee of the Company Board the board of directors of the Company China Merchants China Merchants Expressway Network & Technology Holdings Co Ltd. ( ), a joint stock limited company established in the PRC on December招商局公路網絡科技控股股 18, 1993, whose shares份有限公司 are listed on the Shenzhen Stock Exchange Company or Zhejiang Zhejiang Expressway Co., Ltd., a joint stock limited company incorporated in the PRC with limited Expressway liability on March 1, 1997 Communications Group Zhejiang Communications Investment Group Co., Ltd. ( ), a wholly state-owned enterprise established in the PRC, on December浙江省交通投資集團有限公司 29, 2001 and the controlling shareholder of the Company Connected Person has the meaning ascribed to it under the Listing Rules Controlling Shareholder has the meaning ascribed to it under the Listing Rules De’an Co Deqing County De’an Highway Construction Co., Ltd.