Clinically Relevant Concentrations of Anticancer Drugs: a Guide for Nonclinical Studies Dane R
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Prospective, Multicenter, Phase-II Trial of Ibrutinib Plus Venetoclax In
BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) BMJ Open HOVON 141 CLL Version 4, 20 DEC 2018 A prospective, multicenter, phase-II trial of ibrutinib plus venetoclax in patients with creatinine clearance ≥ 30 ml/min who have relapsed or refractory chronic lymphocytic leukemia (RR-CLL) with or without TP53 aberrations HOVON 141 CLL / VIsion Trial of the HOVON and Nordic CLL study groups PROTOCOL Principal Investigator : Arnon P Kater (HOVON) Carsten U Niemann (Nordic CLL study Group)) Sponsor : HOVON EudraCT number : 2016-002599-29 ; Page 1 of 107 Levin M-D, et al. BMJ Open 2020; 10:e039168. doi: 10.1136/bmjopen-2020-039168 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) BMJ Open Levin M-D, et al. BMJ Open 2020; 10:e039168. doi: 10.1136/bmjopen-2020-039168 BMJ Publishing Group Limited (BMJ) disclaims all liability and responsibility arising from any reliance Supplemental material placed on this supplemental material which has been supplied by the author(s) BMJ Open HOVON 141 CLL Version 4, 20 DEC 2018 LOCAL INVESTIGATOR SIGNATURE PAGE Local site name: Signature of Local Investigator Date Printed Name of Local Investigator By my signature, I agree to personally supervise the conduct of this study in my affiliation and to ensure its conduct in compliance with the protocol, informed consent, IRB/EC procedures, the Declaration of Helsinki, ICH Good Clinical Practices guideline, the EU directive Good Clinical Practice (2001-20-EG), and local regulations governing the conduct of clinical studies. -

Study Protocol
Clovis Oncology, Inc. Clinical Protocol Oral rucaparib (CO-338) CO-338-014 7 July 2016 CONFIDENTIAL A Multicenter, Randomized, Double-Blind, Placebo-Controlled Phase 3 Study of Rucaparib as Switch Maintenance Following Platinum-Based Chemotherapy in Patients with Platinum-Sensitive, High-Grade Serous or Endometrioid Epithelial Ovarian, Primary Peritoneal or Fallopian Tube Cancer Protocol Number: CO-338-014 Investigational Product: Rucaparib (CO-338) Eudra CT Number: IND Number: Development Phase: Phase 3 Indications Studied: Platinum-sensitive, high-grade serous and endometrioid epithelial ovarian, primary peritoneal, and fallopian tube cancer Sponsor Name and Address: Clovis Oncology, Inc. 5500 Flatiron Parkway Suite 100 Boulder, CO 80301 USA Phone Number: 303-625-5000 Facsimile Number: 303-245-0360 Responsible Medical Officer: Compliance Statement: This study will be conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki, clinical research guidelines established by the Code of Federal Regulations (Title 21, CFR Parts 50, 56, and 312), and ICH GCP Guidelines. Essential study documents will be archived in accordance with applicable regulations. CONFIDENTIALITY STATEMENT The information in this document contains commercial information and trade secrets that are privileged or confidential and may not be disclosed unless such disclosure is required by applicable laws and regulations. In any event, persons to whom the information is disclosed must be informed that the information is privileged or confidential and may not be further disclosed by them. These restrictions on disclosure will apply equally to all future information supplied to you which is indicated as privileged or confidential. 1 Clovis Oncology, Inc. Clinical Protocol Oral rucaparib (CO-338) CO-338-014 Coordinating Investigators for the Study Coordinating Investigator for North America: Coordinating Investigator for Europe, Middle East, and Asia Pacific: 2 Clovis Oncology, Inc. -

Modifications on the Basic Skeletons of Vinblastine and Vincristine
Molecules 2012, 17, 5893-5914; doi:10.3390/molecules17055893 OPEN ACCESS molecules ISSN 1420-3049 www.mdpi.com/journal/molecules Review Modifications on the Basic Skeletons of Vinblastine and Vincristine Péter Keglevich, László Hazai, György Kalaus and Csaba Szántay * Department of Organic Chemistry and Technology, University of Technology and Economics, H-1111 Budapest, Szt. Gellért tér 4, Hungary * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel: +36-1-463-1195; Fax: +36-1-463-3297. Received: 30 March 2012; in revised form: 9 May 2012 / Accepted: 10 May 2012 / Published: 18 May 2012 Abstract: The synthetic investigation of biologically active natural compounds serves two main purposes: (i) the total synthesis of alkaloids and their analogues; (ii) modification of the structures for producing more selective, more effective, or less toxic derivatives. In the chemistry of dimeric Vinca alkaloids enormous efforts have been directed towards synthesizing new derivatives of the antitumor agents vinblastine and vincristine so as to obtain novel compounds with improved therapeutic properties. Keywords: antitumor therapy; vinblastine; vincristine; derivatives 1. Introduction Vinblastine (1) and vincristine (2) are dimeric alkaloids (Figure 1) isolated from the Madagaskar periwinkle plant (Catharantus roseus), exhibit significant cytotoxic activity and are used in the antitumor therapy as antineoplastic agents. In the course of cell proliferation they act as inhibitors during the metaphase of the cell cycle and by binding to the microtubules inhibit the development of the mitotic spindle. In tumor cells these agents inhibit the DNA repair and the RNA synthesis mechanisms, blocking the DNA-dependent RNA polymerase. Molecules 2012, 17 5894 Figure 1. -

Multi-Discipline Review/Summary, Clinical, Non-Clinical
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 209115Orig1s000 MULTI-DISCIPLINE REVIEW Summary / Clinical / Non-Clinical NDA/BLA Multi-disciplinary Review and Evaluation NDA 209115 Rubraca (rucaparib) 1 NDA/BLA Multi-disciplinary Review and Evaluation Application Type NDA Application Number(s) 209115 Priority or Standard Priority Submit Date(s) June 23, 2016 Received Date(s) June 23, 2016 PDUFA Goal Date February 23, 2017 Division/Office DOP1/OHOP Review Completion Date November 22, 2016 Established Name Rucaparib (Proposed) Trade Name RUBRACA Pharmacologic Class Poly (ADP-Ribose) Polymerase 1 inhibitor Code name AG-014669, PF-01367338, Applicant Clovis Oncology, Inc. Formulation(s) Oral tablet Dosing Regimen 600 mg PO BID Applicant Proposed Rubraca is indicated as monotherapy treatment of advanced ovarian Indication(s)/Population(s) cancer in patients with deleterious BRCA-mutated tumors, inclusive of both germline BRCA and somatic BRCA mutations (as detected by an FDA approved test), and who have been treated with two or more chemotherapies. Recommendation on Accelerated approval Regulatory Action Recommended Rubraca™ is indicated as monotherapy for the treatment of patients Indication(s)/Population(s) with deleterious BRCA mutation (germline and/or somatic) associated (if applicable) advanced ovarian cancer who have been treated with two or more chemotherapies. Select patients for therapy based on an FDA-approved companion diagnostic for Rubraca. 1-1 Version date: February 1, 2016 for initial rollout (NME/original BLA reviews) Reference ID: 40178304028244 NDA/BLA Multi-disciplinary Review and Evaluation NDA 209115 Rubraca (rucaparib) Contents 1 NDA/BLA Multi-disciplinary Review and Evaluation .......................................................................... 1-1 Reviewers of Multi-Disciplinary Review and Evaluation ........................................................................... -
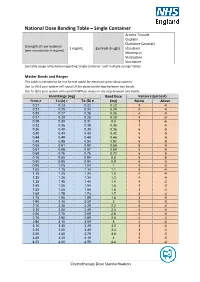
National Dose Banding Table – Single Container
National Dose Banding Table – Single Container Arsenic Trioxide Cisplatin Cladribine (Leustat) Strength of raw material 1 mg/mL Example drug(s) Idarubicin (after reconstitution if required) Mitomycin Vinblastine Vincristine See table usage notes below regarding ‘single container’ and ‘multiple syringe’ tables. Master Bands and Ranges This table is intended to be in a format useful for electronic prescribing systems. Use To (A) if your system will round UP for doses on the step between two bands. Use To (B) if your system will round DOWN for doses on the step between two bands. Band Range (mg) Band Dose Variance (percent) From ≥ To (A) < To (B) ≤ (mg) Below Above 0.21 0.23 0.22 0.22 5 -4 0.23 0.25 0.24 0.24 4 -4 0.25 0.27 0.26 0.26 4 -4 0.27 0.29 0.28 0.28 4 -3 0.29 0.32 0.31 0.3 3 -6 0.32 0.36 0.35 0.34 7 -5 0.36 0.40 0.39 0.38 6 -5 0.40 0.44 0.43 0.42 5 -5 0.44 0.49 0.48 0.46 5 -6 0.49 0.55 0.54 0.52 6 -5 0.55 0.61 0.60 0.58 6 -5 0.61 0.68 0.67 0.64 5 -6 0.68 0.76 0.75 0.72 6 -5 0.76 0.85 0.84 0.8 5 -6 0.85 0.95 0.94 0.9 6 -5 0.95 1.05 1.04 1 5 -5 1.05 1.15 1.14 1.1 5 -4 1.15 1.25 1.24 1.2 4 -4 1.25 1.35 1.34 1.3 4 -4 1.35 1.45 1.44 1.4 4 -3 1.45 1.55 1.54 1.5 4 -3 1.55 1.65 1.64 1.6 3 -3 1.65 1.75 1.74 1.7 3 -3 1.75 1.90 1.89 1.8 3 -5 1.90 2.10 2.09 2 5 -5 2.10 2.30 2.29 2.2 5 -4 2.30 2.50 2.49 2.4 4 -4 2.50 2.70 2.69 2.6 4 -4 2.70 2.90 2.89 2.8 4 -3 2.90 3.10 3.09 3 4 -3 3.10 3.30 3.29 3.2 3 -3 3.30 3.50 3.49 3.4 3 -3 3.50 3.80 3.79 3.6 3 -5 3.80 4.20 4.19 4 5 -5 4.20 4.60 4.59 4.4 5 -4 Chemotherapy Dose Standardisation Band Range (mg) -

Assessment of Overall Survival, Quality of Life, And
Supplementary Online Content Salas-Vega S, Iliopoulos O, Mossialos E. Assesment of overall survival, quality of life, and safety benefits associated with new cancer medicines [published online December 29, 2016]. JAMA Oncol. doi:10.1001/jamaoncol.2016.4166 eTable 1. Therapeutic profile of all cancer medicines approved by the FDA between 2003- 2013 eTable 2. Regulatory evidence in support of classification of drug clinical benefits eFigure 1. Number of cancer drugs that were evaluated by all three HTA agencies, sorted by magnitude of clinical benefits eTable 3. Interagency agreement–Krippendorff’s alpha coefficients eMethods This supplementary material has been provided by the authors to give readers additional information about their work. Online-Only Supplement © 2016 American Medical Association. All rights reserved. Downloaded From: https://jamanetwork.com/ on 09/25/2021 Clinical value of cancer medicines Contents eExhibits ......................................................................................................................................................... 3 eTable 1. Therapeutic profile of all cancer medicines approved by the FDA between 2003- 2013 (Summary of eTable 2) ................................................................................................................... 3 eTable 2. Regulatory evidence in support of classification of drug clinical benefits ....................... 6 eFigure 1. Number of cancer drugs that were evaluated by all three HTA agencies, sorted by magnitude of clinical benefits -

(12) Patent Application Publication (10) Pub. No.: US 2017/0209462 A1 Bilotti Et Al
US 20170209462A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2017/0209462 A1 Bilotti et al. (43) Pub. Date: Jul. 27, 2017 (54) BTK INHIBITOR COMBINATIONS FOR Publication Classification TREATING MULTIPLE MYELOMA (51) Int. Cl. (71) Applicant: Pharmacyclics LLC, Sunnyvale, CA A 6LX 3/573 (2006.01) A69/20 (2006.01) (US) A6IR 9/00 (2006.01) (72) Inventors: Elizabeth Bilotti, Sunnyvale, CA (US); A69/48 (2006.01) Thorsten Graef, Los Altos Hills, CA A 6LX 3/59 (2006.01) (US) A63L/454 (2006.01) (52) U.S. Cl. CPC .......... A61 K3I/573 (2013.01); A61K 3 1/519 (21) Appl. No.: 15/252,385 (2013.01); A61 K3I/454 (2013.01); A61 K 9/0053 (2013.01); A61K 9/48 (2013.01); A61 K (22) Filed: Aug. 31, 2016 9/20 (2013.01) (57) ABSTRACT Disclosed herein are pharmaceutical combinations, dosing Related U.S. Application Data regimen, and methods of administering a combination of a (60) Provisional application No. 62/212.518, filed on Aug. BTK inhibitor (e.g., ibrutinib), an immunomodulatory agent, 31, 2015. and a steroid for the treatment of a hematologic malignancy. US 2017/0209462 A1 Jul. 27, 2017 BTK INHIBITOR COMBINATIONS FOR Subject in need thereof comprising administering pomalido TREATING MULTIPLE MYELOMA mide, ibrutinib, and dexamethasone, wherein pomalido mide, ibrutinib, and dexamethasone are administered con CROSS-REFERENCE TO RELATED currently, simulataneously, and/or co-administered. APPLICATION 0008. In some aspects, provided herein is a method of treating a hematologic malignancy in a subject in need 0001. This application claims the benefit of U.S. -

Vorinostat—An Overview Aditya Kumar Bubna
E-IJD RESIDENTS' PAGE Vorinostat—An Overview Aditya Kumar Bubna Abstract From the Consultant Vorinostat is a new drug used in the management of cutaneous T cell lymphoma when the Dermatologist, Kedar Hospital, disease persists, gets worse or comes back during or after treatment with other medicines. It is Chennai, Tamil Nadu, India an efficacious and well tolerated drug and has been considered a novel drug in the treatment of this condition. Currently apart from cutaneous T cell lymphoma the role of Vorinostat for Address for correspondence: other types of cancers is being investigated both as mono-therapy and combination therapy. Dr. Aditya Kumar Bubna, Kedar Hospital, Mugalivakkam Key Words: Cutaneous T cell lymphoma, histone deacytelase inhibitor, Vorinostat Main Road, Porur, Chennai - 600 125, Tamil Nadu, India. E-mail: [email protected] What was known? • Vorinostat is a histone deacetylase inhibitor. • It is an FDA approved drug for the treatment of cutaneous T cell lymphoma. Introduction of Vorinostat is approximately 9. Vorinostat is slightly Vorinostat is a histone deacetylase (HDAC) inhibitor, soluble in water, alcohol, isopropanol and acetone and is structurally belonging to the hydroxymate group. Other completely soluble in dimethyl sulfoxide. drugs in this group include Givinostat, Abexinostat, Mechanism of action Panobinostat, Belinostat and Trichostatin A. These Vorinostat is a broad inhibitor of HDAC activity and inhibits are an emergency class of drugs with potential anti- class I and class II HDAC enzymes.[2,3] However, Vorinostat neoplastic activity. These drugs were developed with the does not inhibit HDACs belonging to class III. Based on realization that apart from genetic mutation, alteration crystallographic studies, it has been seen that Vorinostat of HDAC enzymes affected the phenotypic and genotypic binds to the zinc atom of the catalytic site of the HDAC expression in cells, which in turn lead to disturbed enzyme with the phenyl ring of Vorinostat projecting out of homeostasis and neoplastic growth. -

The New Therapeutic Strategies in Pediatric T-Cell Acute Lymphoblastic Leukemia
International Journal of Molecular Sciences Review The New Therapeutic Strategies in Pediatric T-Cell Acute Lymphoblastic Leukemia Marta Weronika Lato 1 , Anna Przysucha 1, Sylwia Grosman 1, Joanna Zawitkowska 2 and Monika Lejman 3,* 1 Student Scientific Society, Laboratory of Genetic Diagnostics, Medical University of Lublin, 20-093 Lublin, Poland; [email protected] (M.W.L.); [email protected] (A.P.); [email protected] (S.G.) 2 Department of Pediatric Hematology, Oncology and Transplantology, Medical University of Lublin, 20-093 Lublin, Poland; [email protected] 3 Laboratory of Genetic Diagnostics, Medical University of Lublin, 20-093 Lublin, Poland * Correspondence: [email protected] Abstract: Childhood acute lymphoblastic leukemia is a genetically heterogeneous cancer that ac- counts for 10–15% of T-cell acute lymphoblastic leukemia (T-ALL) cases. The T-ALL event-free survival rate (EFS) is 85%. The evaluation of structural and numerical chromosomal changes is important for a comprehensive biological characterization of T-ALL, but there are currently no ge- netic prognostic markers. Despite chemotherapy regimens, steroids, and allogeneic transplantation, relapse is the main problem in children with T-ALL. Due to the development of high-throughput molecular methods, the ability to define subgroups of T-ALL has significantly improved in the last few years. The profiling of the gene expression of T-ALL has led to the identification of T-ALL subgroups, and it is important in determining prognostic factors and choosing an appropriate treatment. Novel therapies targeting molecular aberrations offer promise in achieving better first remission with the Citation: Lato, M.W.; Przysucha, A.; hope of preventing relapse. -

Cancer Drug Pharmacology Table
CANCER DRUG PHARMACOLOGY TABLE Cytotoxic Chemotherapy Drugs are classified according to the BC Cancer Drug Manual Monographs, unless otherwise specified (see asterisks). Subclassifications are in brackets where applicable. Alkylating Agents have reactive groups (usually alkyl) that attach to Antimetabolites are structural analogues of naturally occurring molecules DNA or RNA, leading to interruption in synthesis of DNA, RNA, or required for DNA and RNA synthesis. When substituted for the natural body proteins. substances, they disrupt DNA and RNA synthesis. bendamustine (nitrogen mustard) azacitidine (pyrimidine analogue) busulfan (alkyl sulfonate) capecitabine (pyrimidine analogue) carboplatin (platinum) cladribine (adenosine analogue) carmustine (nitrosurea) cytarabine (pyrimidine analogue) chlorambucil (nitrogen mustard) fludarabine (purine analogue) cisplatin (platinum) fluorouracil (pyrimidine analogue) cyclophosphamide (nitrogen mustard) gemcitabine (pyrimidine analogue) dacarbazine (triazine) mercaptopurine (purine analogue) estramustine (nitrogen mustard with 17-beta-estradiol) methotrexate (folate analogue) hydroxyurea pralatrexate (folate analogue) ifosfamide (nitrogen mustard) pemetrexed (folate analogue) lomustine (nitrosurea) pentostatin (purine analogue) mechlorethamine (nitrogen mustard) raltitrexed (folate analogue) melphalan (nitrogen mustard) thioguanine (purine analogue) oxaliplatin (platinum) trifluridine-tipiracil (pyrimidine analogue/thymidine phosphorylase procarbazine (triazine) inhibitor) -

Clinical Policy: Pralatrexate (Folotyn)
Clinical Policy: Pralatrexate (Folotyn) Reference Number: CP.PHAR.313 Effective Date: 02.01.17 Last Review Date: 11.19 Coding Implications Line of Business: HIM*, Medicaid, HIM-Medical Benefit Revision Log See Important Reminder at the end of this policy for important regulatory and legal information. Description Pralatrexate injection (Folotyn®) is a folate analog metabolic inhibitor. ____________ *For Health Insurance Marketplace (HIM), if request is through pharmacy benefit, Folotyn (40 mg/2mL vial) is non-formulary and cannot be approved using these criteria; refer to the formulary exception policy, HIM.PA.103. FDA Approved Indication(s) Folotyn is indicated for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL). Policy/Criteria Provider must submit documentation (such as office chart notes, lab results or other clinical information) supporting that member has met all approval criteria. It is the policy of health plans affiliated with Centene Corporation® that Folotyn is medically necessary when the following criteria are met: I. Initial Approval Criteria A. Peripheral T-Cell Lymphoma (must meet all): 1. Diagnosis of PTCL; 2. Prescribed by or in consultation with an oncologist or hematologist; 3. Age ≥ 18 years; 4. Failed prior therapy (see Appendix B for examples); *Prior authorization may be required for prior therapies 5. Request meets one of the following (a or b):* a. Dose does not exceed 30 mg/m2 once weekly for 6 weeks in 7-week cycles; b. Dose is supported by practice guidelines or peer-reviewed literature for the relevant off-label use (prescriber must submit supporting evidence). *Prescribed regimen must be FDA-approved or recommended by NCCN. -

Palifermin (Kepivance™) in the Treatment of Mucositis
Session V • From cell biology to cell therapy Palifermin (Kepivance™) in the treatment of mucositis [haematologica reports] 2005;1(8):41-45 EMMANOUILIDES C n patients undergoing high dose chemo- factor (KGF) is a 28 kD member of the therapy and hematopoietic stem cell fibroblast growth factor family with epithe- Associate Profesor transplant, oral mucositis (OM) is one of lial cell proliferative properties.26 Palifermin of Medicine, UCLA, I Division Haematology-Oncology, the most debilitating and annoying side (Kepivance™) is a truncated, recombinant Thessaloniki, Greece effects. This complication results from form of human keratinocyte growth factor cytotoxic injury to the epithelial lining of (rHuKGF) that has been approved in the the oropharyngeal mucosa, although USA in 2004 to decrease the incidence and lesions of the whole gastrointestinal tract duration of severe OM in patients with also occur.1 The severity of OM varies from hematologic malignancies receiving myelo- erythema and edema accompanied by mild toxic therapy requiring HSCT support. Pal- soreness to full mucosal thickness ulcera- ifermin (recombinant human keratinocyte tions penetrating into the submucosa, growth factor) is an N-terminal, truncated often resulting in severe pain requiring version of endogenous keratinocyte growth narcotic analgesia and impaired swallow- factor with biologic activity similar to that ing, prolonged hospitalization, and of the native protein, but with increased increased risks for infections and poten- stability 26 In animal models of chemother- tially life-threatening sequelae.2,3 Between apy, radiotherapy, and hematopoietic stem- 40% to 80% of cancer patients undergo- cell transplantation27,28 palifermin protected ing intensive treatment regimens requiring several types of epithelial tissues.