Development of Phenotypic Assays for Identifying Novel Blockers of L-Type
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
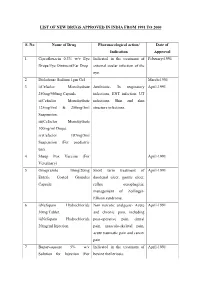
List of New Drugs Approved in India from 1991 to 2000
LIST OF NEW DRUGS APPROVED IN INDIA FROM 1991 TO 2000 S. No Name of Drug Pharmacological action/ Date of Indication Approval 1 Ciprofloxacin 0.3% w/v Eye Indicated in the treatment of February-1991 Drops/Eye Ointment/Ear Drop external ocular infection of the eye. 2 Diclofenac Sodium 1gm Gel March-1991 3 i)Cefaclor Monohydrate Antibiotic- In respiratory April-1991 250mg/500mg Capsule. infections, ENT infection, UT ii)Cefaclor Monohydrate infections, Skin and skin 125mg/5ml & 250mg/5ml structure infections. Suspension. iii)Cefaclor Monohydrate 100mg/ml Drops. iv)Cefaclor 187mg/5ml Suspension (For paediatric use). 4 Sheep Pox Vaccine (For April-1991 Veterinary) 5 Omeprazole 10mg/20mg Short term treatment of April-1991 Enteric Coated Granules duodenal ulcer, gastric ulcer, Capsule reflux oesophagitis, management of Zollinger- Ellison syndrome. 6 i)Nefopam Hydrochloride Non narcotic analgesic- Acute April-1991 30mg Tablet. and chronic pain, including ii)Nefopam Hydrochloride post-operative pain, dental 20mg/ml Injection. pain, musculo-skeletal pain, acute traumatic pain and cancer pain. 7 Buparvaquone 5% w/v Indicated in the treatment of April-1991 Solution for Injection (For bovine theileriosis. Veterinary) 8 i)Kitotifen Fumerate 1mg Anti asthmatic drug- Indicated May-1991 Tablet in prophylactic treatment of ii)Kitotifen Fumerate Syrup bronchial asthma, symptomatic iii)Ketotifen Fumerate Nasal improvement of allergic Drops conditions including rhinitis and conjunctivitis. 9 i)Pefloxacin Mesylate Antibacterial- In the treatment May-1991 Dihydrate 400mg Film Coated of severe infection in adults Tablet caused by sensitive ii)Pefloxacin Mesylate microorganism (gram -ve Dihydrate 400mg/5ml Injection pathogens and staphylococci). iii)Pefloxacin Mesylate Dihydrate 400mg I.V Bottles of 100ml/200ml 10 Ofloxacin 100mg/50ml & Indicated in RTI, UTI, May-1991 200mg/100ml vial Infusion gynaecological infection, skin/soft lesion infection. -

QT Prolongation Due to Roxithromycin
Postgrad Med J 2000;76:651–654 651 Postgrad Med J: first published as 10.1136/pmj.76.900.651 on 1 October 2000. Downloaded from ADVERSE DRUG REACTION QT prolongation due to roxithromycin A Woywodt, U Grommas, W Buth, W RaZenbeul Roxithromycin and other macrolide antimicro- placement of the apex beat, a prominent third bials are widely used for a broad variety of heart sound, coarse rales over both lung fields infections such as upper respiratory tract infec- and pitting oedema of both ankles. The patient tion and community acquired pneumonia. was taken to an intensive care unit. Acute myo- Prolongation of the QT interval, torsade de cardial infarction was ruled out and frusemide pointes polymorphic ventricular tachycardia, was begun intravenously. An electrocardio- and sudden death are well described but little gram (ECG) on admission showed sinus known adverse reactions common to all rhythm and incomplete left bundle branch macrolides. We report the case of a 72 year old block; QT intervals were normal (QT interval patient with congestive heart failure caused by 380 ms, corrected QT interval according to University of ischaemic heart disease who developed severe Bazett’s formula [QTc] 390 ms). Roxithromy- Hannover Medical prolongation of the QT interval after three days cin (Roussel UCLAF, Romainville, France) School, 30623 of treatment with roxithromycin. 150 mg twice a day was initiated for suspected Hannover, Germany: pneumonia. On the third hospital day, he was Department of Nephrology Case report transferred to a general medical ward. A Woywodt A 72 year old man presented with severe On admission there, the patient was gener- W Buth congestive heart failure. -

Dorset Medicines Advisory Group
DORSET CARDIOLOGY WORKING GROUP GUIDELINE FOR CALCIUM CHANNEL BLOCKERS IN HYPERTENSION SUMMARY The pan-Dorset cardiology working group continues to recommend the use of amlodipine (a third generation dihydropyridine calcium-channel blocker) as first choice calcium channel blocker on the pan-Dorset formulary for hypertension. Lercanidipine is second choice, lacidipine third choice and felodipine is fourth choice. This is due to preferable side effect profiles in terms of ankle oedema and relative costs of the preparations. Note: where angina is the primary indication or is a co-morbidity prescribers must check against the specific product characteristics (SPC) for an individual drug to confirm this is a licensed indication. N.B. Lacidipine and lercandipine are only licensed for use in hypertension. Chapter 02.06.02 CCBs section of the Formulary has undergone an evidence-based review. A comprehensive literature search was carried out on NHS Evidence, Medline, EMBASE, Cochrane Database, and UK Duets. This was for recent reviews or meta-analyses on calcium channel blockers from 2009 onwards (comparative efficacy and side effects) and randomised controlled trials (RCTs). REVIEW BACKGROUND Very little good quality evidence exists. No reviews, meta-analyses or RCTs were found covering all calcium channel blockers currently on the formulary. Another limitation was difficulty obtaining full text original papers for some of the references therefore having to use those from more obscure journals instead. Some discrepancies exist between classification of generations of dihydropyridine CCBs, depending upon the year of publication of the reference/authors’ interpretation. Dihydropyridine (DHP) CCBs tend to be more potent vasodilators than non-dihydropyridine (non-DHP) CCBs (diltiazem, verapamil), but the latter have greater inotropic effects. -

Ventricular Arrhythmias Associated with Lidoflazine: Side-Effects Observed in a Randomized Trial •Y
European Heart Journal (1983) 4, 889-893 Ventricular arrhythmias associated with lidoflazine: side-effects observed in a randomized trial •y- S. P. HANLEY AND J. R. HAMPTON Department of Medicine, University Hospital, Queen's Medical Centre, Nottingham NG7 2UH, U.K. Downloaded from https://academic.oup.com/eurheartj/article/4/12/889/503490 by guest on 29 September 2021 KEY WORDS: Angina, exercise testing, lidoflazine, propranolol, ventricular tachycardia. Twenty-four patients received either propranolol, lidoflazine (Clinium), or propranolol/lidoflazine com- binations in a study designed to evaluate the effect of these drugs in angina pectoris. Five patients developed ventricular tachycardia when receiving either lidoflazine or lidoflazine and propranolol in combination; one of these patients died. In addition, one patient died suddenly while being treated with propranolol alone. Lidoflazine therapy was associated with a significant prolongation of the QT interval of the electro- cardiogram. Lidoflazine either alone or in combination with propranolol, appears to induce ventricular tachycardia. Introduction patients already being treated with a beta-blocking, Patients with ischaemic heart disease have an whose symptoms were not adequately controlled; increased risk of death and of developing arrhyth- the second protocol was designed for patients who mias: in any clinical trial of a potential therapeutic were receiving no other treatment other than sub- agent for angina, such events are likely to occur by lingual glyceryl trinitrate. None of thqse patients chance. During a study of lidoflazine we detected was in clinical heart failure. In each of these studies an unacceptable incidence of arrhythmias which exercise tolerance was assessed by treadmill testing led us to discontinue the investigation. -

Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Cr
Drug Name Plate Number Well Location % Inhibition, Screen Axitinib 1 1 20 Gefitinib (ZD1839) 1 2 70 Sorafenib Tosylate 1 3 21 Crizotinib (PF-02341066) 1 4 55 Docetaxel 1 5 98 Anastrozole 1 6 25 Cladribine 1 7 23 Methotrexate 1 8 -187 Letrozole 1 9 65 Entecavir Hydrate 1 10 48 Roxadustat (FG-4592) 1 11 19 Imatinib Mesylate (STI571) 1 12 0 Sunitinib Malate 1 13 34 Vismodegib (GDC-0449) 1 14 64 Paclitaxel 1 15 89 Aprepitant 1 16 94 Decitabine 1 17 -79 Bendamustine HCl 1 18 19 Temozolomide 1 19 -111 Nepafenac 1 20 24 Nintedanib (BIBF 1120) 1 21 -43 Lapatinib (GW-572016) Ditosylate 1 22 88 Temsirolimus (CCI-779, NSC 683864) 1 23 96 Belinostat (PXD101) 1 24 46 Capecitabine 1 25 19 Bicalutamide 1 26 83 Dutasteride 1 27 68 Epirubicin HCl 1 28 -59 Tamoxifen 1 29 30 Rufinamide 1 30 96 Afatinib (BIBW2992) 1 31 -54 Lenalidomide (CC-5013) 1 32 19 Vorinostat (SAHA, MK0683) 1 33 38 Rucaparib (AG-014699,PF-01367338) phosphate1 34 14 Lenvatinib (E7080) 1 35 80 Fulvestrant 1 36 76 Melatonin 1 37 15 Etoposide 1 38 -69 Vincristine sulfate 1 39 61 Posaconazole 1 40 97 Bortezomib (PS-341) 1 41 71 Panobinostat (LBH589) 1 42 41 Entinostat (MS-275) 1 43 26 Cabozantinib (XL184, BMS-907351) 1 44 79 Valproic acid sodium salt (Sodium valproate) 1 45 7 Raltitrexed 1 46 39 Bisoprolol fumarate 1 47 -23 Raloxifene HCl 1 48 97 Agomelatine 1 49 35 Prasugrel 1 50 -24 Bosutinib (SKI-606) 1 51 85 Nilotinib (AMN-107) 1 52 99 Enzastaurin (LY317615) 1 53 -12 Everolimus (RAD001) 1 54 94 Regorafenib (BAY 73-4506) 1 55 24 Thalidomide 1 56 40 Tivozanib (AV-951) 1 57 86 Fludarabine -

Prohibited Substances List
Prohibited Substances List This is the Equine Prohibited Substances List that was voted in at the FEI General Assembly in November 2009 alongside the new Equine Anti-Doping and Controlled Medication Regulations(EADCMR). Neither the List nor the EADCM Regulations are in current usage. Both come into effect on 1 January 2010. The current list of FEI prohibited substances remains in effect until 31 December 2009 and can be found at Annex II Vet Regs (11th edition) Changes in this List : Shaded row means that either removed or allowed at certain limits only SUBSTANCE ACTIVITY Banned Substances 1 Acebutolol Beta blocker 2 Acefylline Bronchodilator 3 Acemetacin NSAID 4 Acenocoumarol Anticoagulant 5 Acetanilid Analgesic/anti-pyretic 6 Acetohexamide Pancreatic stimulant 7 Acetominophen (Paracetamol) Analgesic/anti-pyretic 8 Acetophenazine Antipsychotic 9 Acetylmorphine Narcotic 10 Adinazolam Anxiolytic 11 Adiphenine Anti-spasmodic 12 Adrafinil Stimulant 13 Adrenaline Stimulant 14 Adrenochrome Haemostatic 15 Alclofenac NSAID 16 Alcuronium Muscle relaxant 17 Aldosterone Hormone 18 Alfentanil Narcotic 19 Allopurinol Xanthine oxidase inhibitor (anti-hyperuricaemia) 20 Almotriptan 5 HT agonist (anti-migraine) 21 Alphadolone acetate Neurosteriod 22 Alphaprodine Opiod analgesic 23 Alpidem Anxiolytic 24 Alprazolam Anxiolytic 25 Alprenolol Beta blocker 26 Althesin IV anaesthetic 27 Althiazide Diuretic 28 Altrenogest (in males and gelidngs) Oestrus suppression 29 Alverine Antispasmodic 30 Amantadine Dopaminergic 31 Ambenonium Cholinesterase inhibition 32 Ambucetamide Antispasmodic 33 Amethocaine Local anaesthetic 34 Amfepramone Stimulant 35 Amfetaminil Stimulant 36 Amidephrine Vasoconstrictor 37 Amiloride Diuretic 1 Prohibited Substances List This is the Equine Prohibited Substances List that was voted in at the FEI General Assembly in November 2009 alongside the new Equine Anti-Doping and Controlled Medication Regulations(EADCMR). -

Studies on the Possible Mechanisms of Lidoflazine Arrhythmogenicity
742 lACC Vol 4, No 4 October 1984 742- 7 Studies on the Possible Mechanisms of Lidoflazine Arrhythmogenicity GAD KEREN, MD, DAVID TEPPER, BA, BRENDA BUTLER, BA, WILLIAM MAGUIRE, MD, PHD, HOWARD WILLENS, MD, DENNIS MIURA, MD, PHD , JOHN C. SaMBERG, MD Bronx. New York Lidoftazine is a calcium channel blocking agent that is Dogsalso underwent programmed electrical stimulation effective and safe in the treatment of angina pectoris, while not receiving medications and then after incre but has been reported to be associated with sudden death mental doses of lidoftazine administered intravenously. when administered for the treatment of supraventricular Lidoflazinedid not cause spontaneous ventricular tachy arrhythmias. Studies were performed in dogs to deter cardia and did not lower the threshold of ventricular mine if lidoflazine caused a rise in serum digoxin con tachycardia induction. Combined administration of Ii centration that could cause arrhythmias or if it was di doflazine and digoxin did not facilitate arrhythmia in rectly arrhythmogenic. Dogsreceived chronic injections duction. These studies do not support a digoxin-lido of digoxin and then digoxin in combination with lido f1azine interaction or a direct arrhythmogenic action of ftazine. No increase in digoxin concentration was found. Iidoflazine. Several clinical studies (1-3) have shown the effectiveness Thus. we undertook studies in dogs to test if a digoxin and safety of lidoflazine in the control of angina pectoris. lidoflazine interaction exists and causes a rise in serum di However, in patients with atrial fibril1ation receiving digi goxin levels . Another series of studies used programmed talis therapy and being treated with Iidoflazine to convert electrical stimulation techniques to determine whether suc the supraventricular arrhythmia. -

Medicine for Prevention of and Treatment for Arteriosclerosis and Hypertension
Europäisches Patentamt *EP001604664A1* (19) European Patent Office Office européen des brevets (11) EP 1 604 664 A1 (12) EUROPEAN PATENT APPLICATION published in accordance with Art. 158(3) EPC (43) Date of publication: (51) Int Cl.7: A61K 31/4422, A61K 45/06, 14.12.2005 Bulletin 2005/50 A61P 9/00, A61P 9/10, A61P 43/00, A61P 9/12, (21) Application number: 04706359.9 A61P 13/12 (22) Date of filing: 29.01.2004 (86) International application number: PCT/JP2004/000861 (87) International publication number: WO 2004/067003 (12.08.2004 Gazette 2004/33) (84) Designated Contracting States: • IWAI, Masaru AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Shigenobu-cho, Onsen-gun, HU IE IT LI LU MC NL PT RO SE SI SK TR Ehime 791-0204 (JP) Designated Extension States: • SADA, Toshio, Sankyo Company, Limited AL LT LV MK Tokyo 140-8710 (JP) • MIZUNO, Makoto, Sankyo Company, Limited (30) Priority: 31.01.2003 JP 2003022990 Tokyo 140-8710 (JP) 07.02.2003 JP 2003030830 (74) Representative: Gibson, Christian John Robert (71) Applicant: Sankyo Company, Limited Marks & Clerk Tokyo 103-8426 (JP) 90 Long Acre London WC2E 9RA (GB) (72) Inventors: • HORIUCHI, Masatsugu Onsen-gun, Ehime 791-0204 (JP) (54) MEDICINE FOR PREVENTION OF AND TREATMENT FOR ARTERIOSCLEROSIS AND HYPERTENSION (57) A medicament comprising the following composition: (A) an angiotensin II receptor antagonist selected from the group of a compound having a general formula (I), pharmacologically acceptable esters thereof and pharmacolog- ically acceptable salts thereof (for example, olmesartan medoxomil and the like); EP 1 604 664 A1 Printed by Jouve, 75001 PARIS (FR) (Cont. -

Antioxidant Effects of Calcium Antagonists Rat Myocardial
223 Antioxidant Effects of Calcium Antagonists ['Ii] Rat Myocardial Membrane Lipid Peroxidation Hitoshi Sugawara, Katsuyuki Tobise, and Kenjiro Kikuchi We studied the antioxidant effects of nine calcium antagonists (nisoldipine, benidipine, nilvadipine, felo- dipine, nicardipine; nitrendipine, nifedipine, verapamil, and diltiazem) by means of rat myocardial membrane lipid peroxidation with a nonenzymatic active oxygen-generating system (DHF/FeC13-ADP). The order of antioxidant potency of these agents was nilvadipine > nisoldipine > felodipine > nicardipine > verapamil > benidipine. Their IC50 values (,uM) were 25.1, 28.2, 42.0, 150.0, 266.1, and 420.0, re- spectively. In contrast, nitrendipine, nifedipine, and diltiazem had little inhibitory effect on lipid peroxi- dation.These six calcium antagonists could be divided into four types on the basis of their antioxidant mechanisms. Nilvadipine, nisoldipine, and verapamil, which showed antioxidant effects both before and after the addition of active oxygen, and reduced the dihydroxyfumarate (DHF) auto-oxidation rate, were chain-breaking and preventive antioxidants. Felodipine, which showed antioxidant effects both before and after exposure to active oxygen and increased the DHF auto-oxidation rate, was only a chain-break- ing antioxidant. Nicardipine, which showed an antioxidant effect only before exposure to active oxygen and reduced the DHF auto-oxidation rate, was mainly a preventive antioxidant. Benidipine, which showed an antioxidant effect only before exposure to active oxygen and had no appreciable effect on the DHF auto-oxidation rate, could interrupt the chain reaction of lipid peroxidation at the initial step alone. Although these results suggest that the antioxidant properties of some calcium antagonists may be beneficial clinically in protecting against cellular damage caused by lipid peroxidation, further studies are required to establish the antioxidant effects of these agents in vivo. -
![Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set](https://docslib.b-cdn.net/cover/8870/ehealth-dsi-ehdsi-v2-2-2-or-ehealth-dsi-master-value-set-1028870.webp)
Ehealth DSI [Ehdsi V2.2.2-OR] Ehealth DSI – Master Value Set
MTC eHealth DSI [eHDSI v2.2.2-OR] eHealth DSI – Master Value Set Catalogue Responsible : eHDSI Solution Provider PublishDate : Wed Nov 08 16:16:10 CET 2017 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 1 of 490 MTC Table of Contents epSOSActiveIngredient 4 epSOSAdministrativeGender 148 epSOSAdverseEventType 149 epSOSAllergenNoDrugs 150 epSOSBloodGroup 155 epSOSBloodPressure 156 epSOSCodeNoMedication 157 epSOSCodeProb 158 epSOSConfidentiality 159 epSOSCountry 160 epSOSDisplayLabel 167 epSOSDocumentCode 170 epSOSDoseForm 171 epSOSHealthcareProfessionalRoles 184 epSOSIllnessesandDisorders 186 epSOSLanguage 448 epSOSMedicalDevices 458 epSOSNullFavor 461 epSOSPackage 462 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 2 of 490 MTC epSOSPersonalRelationship 464 epSOSPregnancyInformation 466 epSOSProcedures 467 epSOSReactionAllergy 470 epSOSResolutionOutcome 472 epSOSRoleClass 473 epSOSRouteofAdministration 474 epSOSSections 477 epSOSSeverity 478 epSOSSocialHistory 479 epSOSStatusCode 480 epSOSSubstitutionCode 481 epSOSTelecomAddress 482 epSOSTimingEvent 483 epSOSUnits 484 epSOSUnknownInformation 487 epSOSVaccine 488 © eHealth DSI eHDSI Solution Provider v2.2.2-OR Wed Nov 08 16:16:10 CET 2017 Page 3 of 490 MTC epSOSActiveIngredient epSOSActiveIngredient Value Set ID 1.3.6.1.4.1.12559.11.10.1.3.1.42.24 TRANSLATIONS Code System ID Code System Version Concept Code Description (FSN) 2.16.840.1.113883.6.73 2017-01 A ALIMENTARY TRACT AND METABOLISM 2.16.840.1.113883.6.73 2017-01 -

Benidipine, a Dihydropyridine-Ca2+ Channel Blocker, Increases the Endothelial Differentiation of Endothelial Progenitor Cells in Vitro
1047 Hypertens Res Vol.29 (2006) No.12 p.1047-1054 Original Article Benidipine, a Dihydropyridine-Ca2+ Channel Blocker, Increases the Endothelial Differentiation of Endothelial Progenitor Cells In Vitro Hiroshi ANDO1), Kosuke NAKANISHI2), Mami SHIBATA1), Kazuhide HASEGAWA2), Kozo YAO2), and Hiromasa MIYAJI1) Benidipine is a dihydropyridine-Ca2+ channel blocker used in the treatment of hypertension and angina pec- toris. In the present study, we examined the effects of benidipine on the endothelial differentiation of circu- lating endothelial progenitor cells (EPCs) using an in vitro culture method. Peripheral blood derived mononuclear cells (PBMCs) containing EPCs were isolated from C57BL/6 mice, and then the cells were cul- tured on vitronectin/gelatin-coated slide glasses. After 7 days of culture, endothelial cells differentiated from EPCs were identified as adherent cells with 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine–labeled acetylated low density lipoprotein (DiI-Ac-LDL) uptake and lectin binding under a fluorescent microscope. Incubation of PBMCs for 7 days with benidipine (0.01–1 µmol/l) significantly increased the number of DiI- Ac-LDL+/fluorescein isothiocyanate-lectin (FITC-Lectin)+ cells. Wortmannin, a phosphoinositide-3 kinase (PI3K) inhibitor, selectively attenuated the effect of benidipine on the endothelial differentiation. In addition, benidipine treatment augmented the phosphorylation of Akt, indicating that the PI3K/Akt pathway contrib- uted, at least in part, to the endothelial differentiation induced by benidipine. These results suggest that the treatment with benidipine may increase the endothelial differentiation of circulating EPCs and contribute to endothelial protection, prevention of cardiovascular disease, and/or an improvement of the prognosis after ischemic damage. -

Benidipine Reduces Albuminuria and Plasma Aldosterone in Mild-To-Moderate Stage Chronic Kidney Disease with Albuminuria
Hypertension Research (2011) 34, 268–273 & 2011 The Japanese Society of Hypertension All rights reserved 0916-9636/11 $32.00 www.nature.com/hr ORIGINAL ARTICLE Benidipine reduces albuminuria and plasma aldosterone in mild-to-moderate stage chronic kidney disease with albuminuria Masanori Abe1, Kazuyoshi Okada1, Noriaki Maruyama1, Shiro Matsumoto1, Takashi Maruyama1, Takayuki Fujita1, Koichi Matsumoto1 and Masayoshi Soma1,2 Benidipine inhibits both L- and T-type Ca channels, and has been shown to dilate the efferent arterioles as effectively as the afferent arterioles. In this study, we conducted an open-label and randomized trial to compare the effects of benidipine with those of amlodipine on blood pressure (BP), albuminuria and aldosterone concentration in hypertensive patients with mild-to- moderate stage chronic kidney disease (CKD). Patients with BPX130/80 mm Hg, with estimated glomerular filtration rate (eGFR) of 30–90 ml minÀ1 per 1.73 m2, and with albuminuria430 mg per g creatinine (Cr), despite treatment with the maximum recommended dose of angiotensin II receptor blockers (ARBs) were randomly assigned to two groups. Patients received either of the following two treatment regimens: 2 mg per day benidipine, which was increased up to a dose of 8 mg per day (n¼52), or 2.5 mg per day amlodipine, which was increased up to a dose of 10 mg per day (n¼52). After 6 months of treatment, a significant and comparable reduction in the systolic and diastolic BP was observed in both groups. The decrease in the urinary albumin to Cr ratio in the benidipine group was significantly lower than that in the amlodipine group.