Optimization of Layered Battery Cathode Materials Synthesized Via Spray Pyrolysis Miklos Lengyel Washington University in St
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Pennsylvania State University
The Pennsylvania State University The Graduate School Graduate Program in Nuclear Engineering ASSESSMENT OF RADIATION AWARENESS TRAINING IN IMMERSIVE VIRTUAL ENVIRONMENTS A Dissertation in Nuclear Engineering by Vaughn E. Whisker III © 2008 Vaughn E. Whisker III Submitted in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy May 2008 The dissertation of Vaughn E. Whisker III was reviewed and approved* by the following: Anthony J. Baratta Professor Emeritus of Nuclear Engineering Thesis Advisor Co-chair of Committee C. Frederick Sears Adjunct Professor of Nuclear Engineering Director, Radiation Science and Engineering Center Co-chair of Committee Lawrence E. Hochreiter Professor of Mechanical and Nuclear Engineering Robert M. Edwards Professor of Nuclear Engineering John I. Messner Associate Professor of Architectural Engineering Jack S. Brenizer Professor of Nuclear Engineering Chair of the Nuclear Engineering Program *Signatures are on file in the Graduate School ii ABSTRACT The prospect of new nuclear power plant orders in the near future and the graying of the current workforce create a need to train new personnel faster and better. Immersive virtual reality (VR) may offer a solution to the training challenge. VR technology presented in a CAVE Automatic Virtual Environment (CAVE) provides a high-fidelity, one-to-one scale environment where areas of the power plant can be recreated and virtual radiation environments can be simulated, making it possible to safely expose workers to virtual radiation in the context of the actual work environment. The use of virtual reality for training is supported by many educational theories; constructivism and discovery learning, in particular. Educational theory describes the importance of matching the training to the task. -

Ashlar-Vellum Xenon Spec Sheet 8X11
XENONTM v8 3D Power and finesse without constraints .TM • Updated Interface • True Solid Modeling General Features • Unified Associative Drawing & • Surface Analysis • Direct or Associative 3D Modeling Modeling Tools for Wireframe, Surfaces • Full Photo-realistic Rendering & • ACIS Modeling Kernel from Spatial & Solids Camera-based Animation • 16 Place Floating Point Accuracy TM • Hybrid Mac/Windows, Office/Home License • Vellum Drafting Assistant for • Design Explorer History Tree Intelligent Snaps and Alignments in • 65,000 Hierarchical Layers X,Y, & Z • Associative 2D View Generation with • Unlimited Colors Auxiliary, Section, and Detail Views • Unlimited Number of Objects and Drawing Size • Unique Hybrid Mac/Windows, Office/ • True Associative Dimensioning • Single File Holds Drawings, Models, Assemblies, Home Licensing etc. • Complete Set of Precision Import/ • Bill of Materials • Direct Multi-sheet PDF Creation TM Export Translators Including ACIS SAT, • License includes Graphite v8 • Universal Binary Support for Intel Mac Parasolid, IGES, STEP, DXF, DWG, EPS, AI, • Associative Assembly Tools User Interface and more • Object & Group Properties (CG, • Updated Intuitive Vellum Interface • Class-A NURBS Surface Modeling Volume, Weight, etc.) • Drafting AssistantTM for Automatic Snaps and Alignments • Smart Cursor Changes Shape Indicating Active Tool and Operations • Strokes and Short Cuts Provide Instant Access to Commands • Tear-off and Auto-hide Palettes • Dockable Tools and Palettes • Palettes can be Flipped Vertically or Horizontally -

21 Miscellaneous Companies
Chapter 21 Miscellaneous Companies Space restrictions simply do not permit me to go into the depth of detail I would like on every company that participated in the early days of the CAD industry nor cover numerous in-house systems developed at major automobile and aerospace companies. Readers will have to be satisfied with the brief descriptions included in this chapter and even then, I have only been able to cover what I consider to be the companies that had the biggest impact. There are hundreds if not thousands of companies that at one time marketed engineering design software. Some of the companies described in this chapter offered just software while other provided both hardware and software. While many have changed names, I have decided to list them alphabetically based upon the name they are best been known by along with earlier and subsequent name changes. Adra Systems (Matrix One) Adra Systems was founded in Lowell, Massachusetts in July 1983 by William Mason, who had been at Applicon from 1973 to 1983, most recently as vice president of operations, James Stenzel, who had been vice president of engineering at Hastech, Inc., and Peter Stoupas, who had earlier been a regional sales manager at Adage and had also worked for Applicon. Mason became the president and CEO, Stenzel the vice president of product development and Stoupas the vice president of marketing. Between 1983 and 1986, the company raised $11.6 million of venture funding from a number of firms including American Research & Development, the company that also provided the initial funding for Digital Equipment Corporation. -

Additive Manufacturing: Analysis of the Economic Context and Evaluation of the Indoor Air Quality, with a Total Quality Management Approach
DIPARTIMENTO DI ECONOMIA, SOCIETÀ, POLITICA CORSO DI DOTTORATO DI RICERCA IN Economia, Società, Diritto CURRICULUM Economia e Management CICLO XXXI Additive Manufacturing: analysis of the economic context and evaluation of the indoor air quality, with a Total Quality Management approach SETTORE SCIENTIFICO DISCIPLINARE: SECS-P/13-SCIENZE MERCEOLOGICHE RELATORE DOTTORANDA Chiar.ma Prof.ssa Federica Murmura Dott.ssa Laura Bravi CO TUTOR Ing. Francesco Balducci Anno Accademico 2017/2018 Summary INTRODUCTION CHAPTER 1: ADDITIVE MANUFACTURING: IS IT THE FUTURE? ABSTRACT .......................................................................................................................... 10 1.1 Additive and Subtractive Manufacturing ...................................................................... 10 1.2 The road towards Additive Manufacturing ................................................................... 13 1.2.1 Prehistory of AM .................................................................................................... 14 1.2.2 First attempts to modern AM ................................................................................. 16 1.2.3 The RepRap project ................................................................................................ 19 1.2.4 The Fab@Home project ......................................................................................... 23 1.3 AM today: 3D printing in the digitalization of manufacturing ..................................... 24 1.3.1 The main Additive Manufacturing -
Catia V5 for Mac
1 / 4 Catia V5 For Mac The CAD-embedded FloEFD product is available for PTC Creo, Dassault Systemes CATIA V5, Siemens NX and Solid Edge. A stand-alone version of the FloEFD .... Catia V5. Manual Mac. As recognized, adventure as well as experience just about lesson, amusement, as with ease as arrangement can be gotten by just .... 9 hours ago — ... recent release of 2.0 supporting NX and CATIA models that provided a ... several times over with the Mac, iPod, iPhone and other inventions.. These files directly open in Ashlar-Vellum Cobalt, Xenon and Argon on either Mac or Windows. Alchemy:Adept will also open Catia v5, Pro/E and Siemens NX .... Download CATIA v5R21 Crack Mac Full Version. CATIA v5R21 Crack Full Updated. Catia software, free download - CATIA V5 Import for AutoCAD, CATIA V4 ... Formats Supported · ACIS - SAT, SAB · AutoCAD - DWG, DXF · CATIA V4 - MODEL, SESSION, EXP · CATIA V5 - CATPART, CATPRODUCT, CATDRAWING · CATIA .... 6.3.1, macOS 10.14/10.15, 04/02/2021 ... 2020 download,Catia V5r21,Catia v5 Crack,Catia v5 Download,Solidworks Crack ... For Windows For Mac For Android.. Where can I get help with eDrawings Publisher for CATIA, Autodesk Inventor, SketchUp, ... From where can I download eDrawings Viewer for Mac? You can .... Mutaz.net. Windows · Android · Mac · Laptops · Firmwares · E-Books · Courses · Blog · Contact Me · Windows Android Mac Laptops Firmwares E-Books Courses .... The Keyzy software suite is SaaS, Mac, and Windows software. Tokens can be ... This video presents a brief installation process of catia V5 R2014. So the .... Dec 10, 2007 — Im at university with my macbook, and Catia V5 has become available for purchase at student rates. -

Vlsi Cad Engineering Grace Gao, Principle Engineer, Rambus Inc
VLSI CAD ENGINEERING GRACE GAO, PRINCIPLE ENGINEER, RAMBUS INC. AUGUST 5, 2017 Agenda • CAD (Computer-Aided Design) ◦ General CAD • CAD innovation over the years (Short Video) ◦ VLSI CAD (EDA) • EDA: Where Electronic Begins (Short Video) • Zoom Into a Microchip (Short Video) • Introduction to Electronic Design Automation ◦ Overview of VLSI Design Cycle ◦ VLSI Manufacturing • Intel: The Making of a Chip with 22nm/3D (Short Video) ◦ EDA Challenges and Future Trend • VLSI CAD Engineering ◦ EDA Vendors and Tools Development ◦ Foundry PDK and IP Reuse ◦ CAD Design Enablement ◦ CAD as Career • Q&A CAD (Computer-Aided Design) General CAD • Computer-aided design (CAD) is the use of computer systems (or workstations) to aid in the creation, modification, analysis, or optimization of a design CAD innovation over the years (Short Video) • https://www.youtube.com/watch?v=ZgQD95NhbXk CAD Tools • Commercial • Freeware and open source Autodesk AutoCAD CAD International RealCAD 123D Autodesk Inventor Bricsys BricsCAD LibreCAD Dassault CATIA Dassault SolidWorks FreeCAD Kubotek KeyCreator Siemens NX BRL-CAD Siemens Solid Edge PTC PTC Creo (formerly known as Pro/ENGINEER) OpenSCAD Trimble SketchUp AgiliCity Modelur NanoCAD TurboCAD IronCAD QCad MEDUSA • ProgeCAD CAD Kernels SpaceClaim PunchCAD Parasolid by Siemens Rhinoceros 3D ACIS by Spatial VariCAD VectorWorks ShapeManager by Autodesk Cobalt Gravotech Type3 Open CASCADE RoutCad RoutCad SketchUp C3D by C3D Labs VLSI CAD (EDA) • Very-large-scale integration (VLSI) is the process of creating an integrated circuit (IC) by combining hundreds of thousands of transistors into a single chip. • The design of VLSI circuits is a major challenge. Consequently, it is impossible to solely rely on manual design approaches. -

Intuitive Motion Simulation for 3D Modeling.™
Intuitive motion simulation for 3D modeling.™ Kinetics 3D Motion Simulation Ashlar-Vellum Kinetics offers an easy-to-use set of precision motion • Optimized product development simulation tools at an affordable price point that’s independent and reduce costs: from any particular CAD or 3D modeling software. Available for both • Intuitive time-based motion Macintosh and Windows, Kinetics enhances engineering design, studies. optimizes product and assembly performance, and reduces the • Use your choice of 3D modeling number of physical prototypes needed. program on Mac or Windows. In its virtual test environment, Kinetics determines the physical • Unique Organic Workflow allows movement of a model or assembly, calculates the forces and testing at any point within the analyzes components, helping to solve common challenges through design. time-based motion studies. • Animate individual components or Simply load the model, appoint the joints (connections), click the entire assemblies. button and watch what happens as it simulates the motion and • Simulate properties of motion. presents an analysis of your model. Unlike other motion simulation • Photo-realistic rendering in real software, Kinetics follows the Ashlar-Vellum Organic Workflow™ time. that allows you to test parts and assemblies at any time or from any • Export to a number of 3D formats. model in your design process. Simulate motions in Kinetics—define time-based, rigid body studies using kinematics or dynamics. Determine motion optimization by evaluating actuator force and joint loads. Assess product performance through it’s cycle of operation. Analyze the design for range of motion, part displacements and overall performance. Works with files from all leading modelers— Kinetics accepts precision (analytical) and approximate (polygonal) models from other leading software including: • Ashlar-Vellum Cobalt™, Xenon™ or Argon™ • Dassault Systems SolidWorks • Siemen’s Solid Edge Gear Assembly modeled by Jesse Luis. -

Turbocad Pro 17 (Competitive Upgrade)
TurboCAD Pro 17 (Competitive Upgrade) TurboCAD Pro 17 is advanced CAD software with comprehensive 2D drafting and detailing tools, powerful, 3D surface and solid modelling, and photorealistic lighting and rendering. Complete Set of 2D/3D Drafting, Design, and Modelling Tools Perfect for professionals including engineers, architects, technical designers etc. Powerful tools for 2D or 3D design including 2D parametric constraints, 3D solid and surface modelling, robust photorealistic rendering ray tracing, lighting and materials, and extensive CAD and graphics file interoperability. Advances productivity with tools that allow for design, modification, presentation, and documentation in an integrated fashion. Excellent workflow to and from other applications to ease collaboration. Open, insert, or embed up to 35 file formats and export up to 28, including .DWG, .DXF, .SKP (Google™ SketchUp™), etc. Plus, reads and writes .DWG and .DXF files from R14 Competitive Upgrade Price: £469 inc.VAT through 2010 including AutoCAD® Architecture Product RRP: £895 inc. VAT extensions. Extensible features with plug-ins, upgrades “TurboCAD is undoubtedly the and an SDK for those wishing to extend most powerful CAD program functionality in a custom manner. available” “not only a remarkable business tool, IMSI/Design, LLC, is the global leader in retail but the software that I have come to CAD (Computer Aided Design). IMSI/Design know as my trusted business products include the award-winning TurboCAD®, partner. TurboCAD's 3D capabilities TurboFLOORPLAN™, TurboSketch™, and are second to none.” DesignCAD™ families of precision design applications. Source: Customer Profiles available for download TurboCAD Pro 17 (Competitive Upgrade) If you already own another CAD or graphic design product, then you can benefit from the full power and productivity of TurboCAD® 17 at a special price – it’s a great way to add the latest in precision 2D/3D design power to your current CAD library. -

July 1994 • Kevsolutlons • 5 F¥ - " ~ Kml.](Ljit.]:~1 the Magazine for CADKEY & Datacad T.~ KEVTALK J~ P.O
LET'S FACE IT! If you use CADI<EY and need a CAM program to machine surfaces at a price you can afford, you need ALL the facts and CUTTING EDGE SURFACES! ,CUTTING EDGE SURFACES has: No oth~r CAM p-ackage has: • all the geometry of CADKEY. • 1 00% CADKEY data base compatibility • surface modeling of FASTSURF • 100% CAOKEY human interface • complete tool path editing • a complete COE & CADL environment • a library of over: 100 post pr:o,cessors • generaLNURBS surface machining • CADKEY IGES cap-abilities CUTIING EDGE SURFACES can be used as a stand alone CAM system for 3-axis milling, drilling, boring, reaming, slotting, pocketing, tapping and contouring Immediate verification of the tool path before cutting reduces material waste. CUTIING EDGE SURFACES is fu lly integrated into Cad key's desktop engineering tools. Now you can manufacture your designs directly from CADKEY part files with I 00% failure-free data transfer. NO TRANSlATION needed! You can also transfer other CAD files using IGES, DXF, and CADL CUTIING EDGE SURFACES offers ease of use unparalleled in the CAD/CAM world, an outstanding customization environment, general surface machining, and the ability to bring surfaces from any CAD system that supports IGES. If you don't have a blue print or a CAD design, we can help with that too. Ask about the reverse engineering abili ty of CADDlnspector. Call your CUTTING EDGE dealer or Cutting Edge Technologies, Inc. c::J 4 Griffin Road North Windsor, CT 06095 • (203) 298-6433 Fax: (203) 298-6490 24 Hr. Automatic Fax Inlo 1-8()().n9-0202 Code 124408 like Reaching Inlo Ihe SCl'een and Moving Design Pal'ls Dil'eelly. -

Understanding the Linux Kernel
Understanding the Linux Kernel Daniel P. Bovet Marco Cesati Publisher: O'Reilly First Edition October 2000 ISBN: 0-596-00002-2, 702 pages Understanding the Linux Kernel helps readers understand how Linux performs best and how it meets the challenge of different environments. The authors introduce each topic by explaining its importance, and show how kernel operations relate to the utilities that are familiar to Unix programmers and users. Table of Contents Preface .......................................................... 1 The Audience for This Book .......................................... 1 Organization of the Material .......................................... 1 Overview of the Book .............................................. 3 Background Information ............................................. 4 Conventions in This Book ........................................... 4 How to Contact Us ................................................. 4 Acknowledgments ................................................. 5 1. Introduction .................................................... 6 1.1 Linux Versus Other Unix-Like Kernels ............................... 6 1.2 Hardware Dependency .......................................... 10 1.3 Linux Versions ................................................ 11 1.4 Basic Operating System Concepts .................................. 12 1.5 An Overview of the Unix Filesystem ................................ 16 1.6 An Overview of Unix Kernels ..................................... 22 2. Memory Addressing ............................................ -
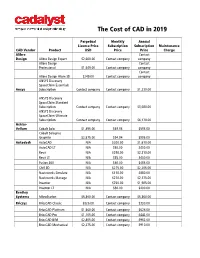
The Cost of CAD in 2019
The Cost of CAD in 2019 Perpetual Monthly Annual License Price Subscription Subscription Maintenance CAD Vendor Product USD Price Price Charge Alibre Contact Design Alibre Design Expert $2,600.00 Contact company company Alibre Design Contact Professional $1,549.00 Contact company company Contact Alibre Design Atom 3D $249.00 Contact company company ANSYS Discovery SpaceClaim Essentials Ansys Subscription Contact company Contact company $1,230.00 ANSYS Discovery SpaceClaim Standard Subscription Contact company Contact company $3,080.00 ANSYS Discovery SpaceClaim Ultimate Subscription Contact company Contact company $6,170.00 Ashlar- Vellum Cobalt Solo $1,495.00 $59.95 $595.00 Cobalt Solo plus Graphite $2,575.00 $94.94 $995.00 Autodesk AutoCAD N/A $200.00 $1,610.00 AutoCAD LT N/A $50.00 $400.00 Revit N/A $290.00 $2,310.00 Revit LT N/A $55.00 $430.00 Fusion 360 N/A $60.00 $495.00 Civil 3D N/A $275.00 $2,205.00 Navisworks Simulate N/A $110.00 $880.00 Navisworks Manage N/A $270.00 $2,175.00 Inventor N/A $250.00 $1,985.00 Inventor LT N/A $50.00 $400.00 Bentley Systems MicroStation $5,360.00 Contact company $5,360.00 Bricsys BricsCAD Classic $826.00 Contact company $330.00 BricsCAD Platinum $1,560.00 Contact company $624.00 BricsCAD Pro $1,105.00 Contact company $442.00 BricsCAD BIM $2,405.00 Contact company $962.00 BricsCAD Mechanical $2,275.00 Contact company $910.00 The Cost of CAD in 2019 Perpetual Monthly Annual License Price Subscription Subscription Maintenance CAD Vendor Product USD Price Price Charge Corel Contact Corporation Corel -

3D Modeling: Thing's I've Learned Computer Modeling Is a Unique
3D modeling: thing's I've learned computer modeling is a unique language with its own inconsistent and broken grammar. It is not intuitive. Keyboard and mouse shortcuts are absolutely neccessary to do many critical functions, and these must be memorized. Different programs do different things well, and I need to switch between programs to make a complex model the way I want to. Rhino: surface modeling and nurbs: good for curvy surfaces. Just starting to learn it, and it's friendlier than cobalt. Heard the dolphin tutorial online is good. I'm gonna try it. Inventor: Heard it's a good alternative to Cobalt. Gonna check it out. Cobalt: shitty interface, but precise and fairly "powerful." uses "point cloud" modeling, which is an infinite number of points in space to define a solid. This is good because it means cobalt understands the relationships between solids in space. this is not true of rhino; if you put a block inside a block in rhino, it does not know that they intersect until you draw a face that connects them. cobalt does know this kind of thing. if you need to intersect a line or plane with a solid, cobalt can do it. interface: very hard to rotate things and there is often no dynamic tool. e.g. you can't freehand a rotation: it must be numerically defined, or reference off of a pre-drawn guide like a line or cube. But good solid modeler. If you're trying to engineer/design an assembly or many parts, this is handy.