Inhibiting Phosphorylation and Aggregation of Tau Protein Using R Domain Peptide Mimetics a Dissertation Presented to the Facul
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Family Name Files That Can Be Accessed in the Boerne Public Library Local and Family History Archives
FAMILY NAME FILES THAT CAN BE ACCESSED IN THE BOERNE PUBLIC LIBRARY LOCAL AND FAMILY HISTORY ARCHIVES A Abadie, Ables, Abshire Ackerman Adair, Adam, Adamek, Adamietz, Adams, Adamson, Adcock, Adkins, Adler, Adrian, Adriance, Agold, Aguirre, Ahr, Ahrlett, Alba, Albrecht, Albright, Alcorn, Alderman, Aldridge, Alexander, Alf, Alford, Algueseva, Allamon, Allen, Allison, Altgelt, Alvarez, Amason, Ammann, Ames, Anchondo, Anderson, Angell, Angle, Ansaldo, Anstiss, Appelt, Armendarez, Armstrong, Arnold, Arnott, Artz, Ashcroft, Asher, Atencio, Atkinson, Aue, Austin, Aycock, B Backer, Bacon, Bacorn, Bain, Baker, Baldwin, Ball, Balser, Bangert, Bankier, Banks, Bannister, Barbaree, Barker, Barkley, Barnard, Barnes, Barnett, Barnette, Barnhart, Baron, Barrett, Barrington, Barron, Barrows, Barta, Barteau, Bartel, Bartels, Barthlow, Barton, Basham, Bates, Batha, Bauer, Baum, Baumann, Bausch, Baxter, Bayard, Bayless, Baynton, Beagle, Bealor, Beam, Bean, Beardslee, Beasley, Beath, Beatty, Beauford, Beaver, Bechtold, Beck, Becker, Beckett, Beckley, Bedford, Bedgood, Bednar, Bedwell, Beem, Beene, Beer, Begia, Behr, Beissner, Bell, Below, Bemus, Benavides, Bender, Bennack, Benefield, Benner, Bennett, Benson, Bentley, Berger, Bergmann, Berlin, Berline, Bernal, Berne, Bernert, Bernhard, Bernstein, Berry, Besch, Beseler, Beshea, Besser, Best, Bettison, Beutnagel, Bevers, Bey, Beyer, Bickel, Bidus, Bien, Bierman, Bierschwale, Bigger, Biggs, Billingsley, Birdsong, Birkner, Bish, Bitzkie, Black, Blackburn, Blackford, Blackwell, Blair, Blaize, Blake, Blakey, Blalock, -

The German Surname Atlas Project ± Computer-Based Surname Geography Kathrin Dräger Mirjam Schmuck Germany
Kathrin Dräger, Mirjam Schmuck, Germany 319 The German Surname Atlas Project ± Computer-Based Surname Geography Kathrin Dräger Mirjam Schmuck Germany Abstract The German Surname Atlas (Deutscher Familiennamenatlas, DFA) project is presented below. The surname maps are based on German fixed network telephone lines (in 2005) with German postal districts as graticules. In our project, we use this data to explore the areal variation in lexical (e.g., Schröder/Schneider µtailor¶) as well as phonological (e.g., Hauser/Häuser/Heuser) and morphological (e.g., patronyms such as Petersen/Peters/Peter) aspects of German surnames. German surnames emerged quite early on and preserve linguistic material which is up to 900 years old. This enables us to draw conclusions from today¶s areal distribution, e.g., on medieval dialect variation, writing traditions and cultural life. Containing not only German surnames but also foreign names, our huge database opens up possibilities for new areas of research, such as surnames and migration. Due to the close contact with Slavonic languages (original Slavonic population in the east, former eastern territories, migration), original Slavonic surnames make up the largest part of the foreign names (e.g., ±ski 16,386 types/293,474 tokens). Various adaptations from Slavonic to German and vice versa occurred. These included graphical (e.g., Dobschinski < Dobrzynski) as well as morphological adaptations (hybrid forms: e.g., Fuhrmanski) and folk-etymological reinterpretations (e.g., Rehsack < Czech Reåak). *** 1. The German surname system In the German speech area, people generally started to use an addition to their given names from the eleventh to the sixteenth century, some even later. -

Magazin Wirtschaft
www.stuttgart.ihk.de 07.2013 Stuttgart - Böblingen - Esslingen-Nürtingen - Göppingen - Ludwigsburg - Rems-Murr Magazin Wirtschaft Ein Service der IHK für Unternehmen in der Region Stuttgart Fachkräfte finden zwischen Alb und Gäu Seite 6 wer hat schon ein ganzes kabinett nur für diewirtschaft? Selbstverständlich wir. Damit die Betriebe gute Rahmenbedingungen haben, sind in unseren Gremien ausschließlich Wirtschaftexperten vertreten. www.stuttgart.ihk.de oder Infoline 0711 2005-0 EDITORIAL Handelskriege kennen keine Gewinner Ähnlich dem Frühjahrswetter in diesem weit kommen? China ist die zweitgrößte Wirt- Jahr ziehen nun auch Wolken über dem schaftsmacht der Welt. Daher sollte das Land Parkett des internationalen Handels beginnen, sich an internationale Spielregeln auf. Auslöser ist die vorläufige Erhebung von zu halten. Globalisierung benötigt faire Wett- EU-Strafzöllen auf chinesische Solarmodule. bewerbsbedingungen, an die sich China zu- China kontert mit einem Anti-Dumping-Ver- weilen jedoch nicht hält. Belegte Fälle von fahren gegen europäischen Wein. Immer Wettbewerbsverstößen gibt es genug. mehr Produkte treten in den Fokus der Dum- ping-Diskussion. Vernünftige Handelspolitik Die EU muss schnellstens sieht anders aus. Mangels Deeskalation wird Verhandlungen aufnehmen die Situation für China und Europa immer tassilo zywietz unangenehmer. Ein Handelskrieg droht. Europa darf davor die Augen nicht ver- Geschäftsführer International Globalisierung erfordert jedoch gegensei- schließen. Nachgiebigkeit ist keine Erfolg ver- der IHK Region Stuttgart tige Rücksichtnahme und Handeln im Sinne sprechende Strategie – damit würde die EU der immer stärker miteinander verbundenen erpressbar. Lässt man China gewähren, kann Volkswirtschaften: China und die EU haben die Methode künftig in der Chemiebranche letztes Jahr Waren und Dienstleistungen in und irgendwann im Autobau angewendet Höhe von über 400 Milliarden Euro ausge- werden. -

Family Group Sheets Surname Index
PASSAIC COUNTY HISTORICAL SOCIETY FAMILY GROUP SHEETS SURNAME INDEX This collection of 660 folders contains over 50,000 family group sheets of families that resided in Passaic and Bergen Counties. These sheets were prepared by volunteers using the Societies various collections of church, ceme tery and bible records as well as city directo ries, county history books, newspaper abstracts and the Mattie Bowman manuscript collection. Example of a typical Family Group Sheet from the collection. PASSAIC COUNTY HISTORICAL SOCIETY FAMILY GROUP SHEETS — SURNAME INDEX A Aldous Anderson Arndt Aartse Aldrich Anderton Arnot Abbott Alenson Andolina Aronsohn Abeel Alesbrook Andreasen Arquhart Abel Alesso Andrews Arrayo Aber Alexander Andriesse (see Anderson) Arrowsmith Abers Alexandra Andruss Arthur Abildgaard Alfano Angell Arthurs Abraham Alje (see Alyea) Anger Aruesman Abrams Aljea (see Alyea) Angland Asbell Abrash Alji (see Alyea) Angle Ash Ack Allabough Anglehart Ashbee Acker Allee Anglin Ashbey Ackerman Allen Angotti Ashe Ackerson Allenan Angus Ashfield Ackert Aller Annan Ashley Acton Allerman Anners Ashman Adair Allibone Anness Ashton Adams Alliegro Annin Ashworth Adamson Allington Anson Asper Adcroft Alliot Anthony Aspinwall Addy Allison Anton Astin Adelman Allman Antoniou Astley Adolf Allmen Apel Astwood Adrian Allyton Appel Atchison Aesben Almgren Apple Ateroft Agar Almond Applebee Atha Ager Alois Applegate Atherly Agnew Alpart Appleton Atherson Ahnert Alper Apsley Atherton Aiken Alsheimer Arbuthnot Atkins Aikman Alterman Archbold Atkinson Aimone -

Surname Folders.Pdf
SURNAME Where Filed Aaron Filed under "A" Misc folder Andrick Abdon Filed under "A" Misc folder Angeny Abel Anger Filed under "A" Misc folder Aberts Angst Filed under "A" Misc folder Abram Angstadt Achey Ankrum Filed under "A" Misc folder Acker Anns Ackerman Annveg Filed under “A” Misc folder Adair Ansel Adam Anspach Adams Anthony Addleman Appenzeller Ader Filed under "A" Misc folder Apple/Appel Adkins Filed under "A" Misc folder Applebach Aduddell Filed under “A” Misc folder Appleman Aeder Appler Ainsworth Apps/Upps Filed under "A" Misc folder Aitken Filed under "A" Misc folder Apt Filed under "A" Misc folder Akers Filed under "A" Misc folder Arbogast Filed under "A" Misc folder Albaugh Filed under "A" Misc folder Archer Filed under "A" Misc folder Alberson Filed under “A” Misc folder Arment Albert Armentrout Albight/Albrecht Armistead Alcorn Armitradinge Alden Filed under "A" Misc folder Armour Alderfer Armstrong Alexander Arndt Alger Arnold Allebach Arnsberger Filed under "A" Misc folder Alleman Arrel Filed under "A" Misc folder Allen Arritt/Erret Filed under “A” Misc folder Allender Filed under "A" Misc folder Aschliman/Ashelman Allgyer Ash Filed under “A” Misc folder Allison Ashenfelter Filed under "A" Misc folder Allumbaugh Filed under "A" Misc folder Ashoff Alspach Asper Filed under "A" Misc folder Alstadt Aspinwall Filed under "A" Misc folder Alt Aston Filed under "A" Misc folder Alter Atiyeh Filed under "A" Misc folder Althaus Atkins Filed under "A" Misc folder Altland Atkinson Alwine Atticks Amalong Atwell Amborn Filed under -

Sales Report
Sales Listing Report Page 1 of 309 McHenry County 05/04/2020 09:46:13 01/01/2019 - 12/31/2019 Township: GRAFTON TWP Document Number Sale Year Sale Type Valid Sale Sale Date Dept. Study Selling Price Parcel Number Built Year Property Type Prop. Class Acres Square Ft. Lot Size Grantor Name Grantee Name Site Address 2019R0006019 2019 Not advertised on mN 02/19/2019 N $115,000.00 WILLIAM MCDONALD PETER J. GODLEWSKI DOLORESS MCDONALD 78 S HEATHER DR 18-01-101-013 0 GARAGE/ NO O O 0040 .00 0 CRYSTAL LAKE, IL 600145018 78 S HEATHER DR CRYSTAL LAKE, IL 60014 -5018 Legal Description: DOC 2019R0006019 LT 16 & 17 BLK 15 R A CEPEKS CRYSTAL VISTA 2019R0027941 2019 Not advertised on mN 07/08/2019 N $150,000.00 CHARLES G. ALEMAN REINVEST HOMES LLC, AN ILLINOIS LIMIT DAWN S. ALEMAN 503 E ALGONQUIN RD 18-01-101-027 0 GARAGE/ NO O O 0040 .00 0 ALGONQUIN, IL 601023004 160 HEATHER DR CRYSTAL LAKE, IL 60014 - Legal Description: DOC 2019R0027941 LT 31 BLK 15 R A CEPEKS CRYSTAL VISTA 2019R0026844 2019 Warranty Deed Y 08/14/2019 Y $172,000.00 THOMAS M. COFFMAN JESSICA N. NICHOLAS KATRINA COFFMAN 1350 THORNWOOD LN 18-01-102-035 0 LDG SINGLE FAM 0040 .00 0 CRYSTAL LAKE, IL 600145042 1350 THORNWOOD LN CRYSTAL LAKE, IL 60014 -5042 Legal Description: DOC 2019R0026844 LT 6 & PT LT 19 LYING N OF & ADJ BLK 14 R A CEPEKS CRYSTAL VISTA 2019R0010406 2019 Warranty Deed Y 04/12/2019 Y $174,000.00 JEREMY M. -

Zion Lutheran's Surname List
Zion Lutheran Church Records 1861–1961, Belleville, Illinois—surnames of families found in this book. © 2011 St. Clair County Genealogical Society, P.O. Box 431, Belleville, IL 62222-0431. Early records of this church—translated from German—include 4200 baptisms with parents named, 2700 confirmation entries, 1268 marriages, 1360 burials, witnesses and sponsors. A CDRom publication is planned by the SCCGS. Interested in a copy? Contact the Society via one of the website www.stclair-ilgs.org links posted there. _EIMAND ANDRECK BAEHR BARTHELHEIMER BEIRAND BETTERTSCH ABEL ANDREGG BAETTGER BARTLING BEISER BETZ ABENDROTH ANDRES BAEUERLE BARTON BEISSWINGERT BEUDA ABERLE ANDREW BAEUMER BARTS BEITERMANN BEURMANN ABRAHAMSON ANDREWS BAGNAUER BARTTELBORT BEITHAUS BEUSE ACHISON ANDRO BAGNET BARTZ BELCOUR BEUTENBACH ACHS ANDRUSHAT BAGSHAW BATER BELINSKI BEUTNAGEL ACKER ANNA BAHORCE BATMAN BELKER BEVIART ACKERMANN ANSTETTE BAHORICH BATRIE BELKERS BEWART ACOEN ANTEROPP BAHR BATSSTE BELL BEYER ADAMS ANTHES BAIER BATTELBORD BELLEVILLE BEYERLEY ADAMSON ANTHONY BAIL BATTOE BELLOFF BICKEL ADELMANN ANTON BAILEY BAUCHER BELMKE BIEBEL ADEN APEL BAKER BAUER BENDE BIEBER ADKINS APPERSON BALAICH BAUMAN BENDER BIEBN ADLER AREY BALARCH BAUMANN BENDIN BIEDERMANN ADRIAN ARING BALARICK BAUMGARTEN BENDINGER BIEHL AGLES ARL BALDEN BAUMGARTNER BENEDICK BIEHLHORN AGNE ARMANNO BALDWIN BAUMUNCH BENEDICT BIEN AHLEMEYER ARMANNS BALKE BAYER BENING BIERERLY AHLERS ARMBRUST BALL BAZ BENNIKE BIERLENBACH AHLERSMEIER ARMBRUSTER BALLHAUS BEAN BENNING BIERMANN AHLERSMEYER ARMENO BALSEKER -
Marriage Certificates
GROOM LAST NAME GROOM FIRST NAME BRIDE LAST NAME BRIDE FIRST NAME DATE PLACE Abbott Calvin Smerdon Dalkey Irene Mae Davies 8/22/1926 Batavia Abbott George William Winslow Genevieve M. 4/6/1920Alabama Abbotte Consalato Debale Angeline 10/01/192 Batavia Abell John P. Gilfillaus(?) Eleanor Rose 6/4/1928South Byron Abrahamson Henry Paul Fullerton Juanita Blanche 10/1/1931 Batavia Abrams Albert Skye Berusha 4/17/1916Akron, Erie Co. Acheson Harry Queal Margaret Laura 7/21/1933Batavia Acheson Herbert Robert Mcarthy Lydia Elizabeth 8/22/1934 Batavia Acker Clarence Merton Lathrop Fannie Irene 3/23/1929East Bethany Acker George Joseph Fulbrook Dorothy Elizabeth 5/4/1935 Batavia Ackerman Charles Marshall Brumsted Isabel Sara 9/7/1917 Batavia Ackerson Elmer Schwartz Elizabeth M. 2/26/1908Le Roy Ackerson Glen D. Mills Marjorie E. 02/06/1913 Oakfield Ackerson Raymond George Sherman Eleanora E. Amelia 10/25/1927 Batavia Ackert Daniel H. Fisher Catherine M. 08/08/1916 Oakfield Ackley Irving Amos Reid Elizabeth Helen 03/17/1926 Le Roy Acquisto Paul V. Happ Elsie L. 8/27/1925Niagara Falls, Niagara Co. Acton Robert Edward Derr Faith Emma 6/14/1913Brockport, Monroe Co. Adamowicz Ian Kizewicz Joseta 5/14/1917Batavia Adams Charles F. Morton Blanche C. 4/30/1908Le Roy Adams Edward Vice Jane 4/20/1908Batavia Adams Edward Albert Considine Mary 4/6/1920Batavia Adams Elmer Burrows Elsie M. 6/6/1911East Pembroke Adams Frank Leslie Miller Myrtle M. 02/22/1922 Brockport, Monroe Co. Adams George Lester Rebman Florence Evelyn 10/21/1926 Corfu Adams John Benjamin Ford Ada Edith 5/19/1920Batavia Adams Joseph Lawrence Fulton Mary Isabel 5/21/1927Batavia Adams Lawrence Leonard Boyd Amy Lillian 03/02/1918 Le Roy Adams Newton B. -
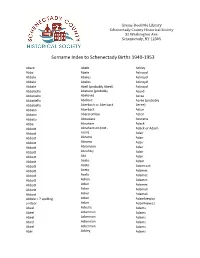
Surname Index to Schenectady Births 1940-1953
Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Abare Abele Ackley Abba Abele Ackroyd Abbale Abeles Ackroyd Abbale Abeles Ackroyd Abbale Abell (probably Abeel) Ackroyd Abbatiello Abelone (probably Acord Abbatiello Abelove) Acree Abbatiello Abelove Acree (probably Abbatiello Aberbach or Aberback Aeree) Abbato Aberback Acton Abbato Abercrombie Acton Abbato Aboudara Acucena Abbe Abraham Adack Abbott Abrahamson (not - Adack or Adach Abbott nson) Adair Abbott Abrams Adair Abbott Abrams Adair Abbott Abramson Adair Abbott Abrofsky Adair Abbott Abt Adair Abbott Aceto Adam Abbott Aceto Adamczak Abbott Aceto Adamec Abbott Aceto Adamec Abbott Acken Adamec Abbott Acker Adamec Abbott Acker Adamek Abbott Acker Adamek Abbzle = ? spelling Acker Adamkiewicz unclear Acker Adamkiewicz Abeel Ackerle Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abeel Ackerman Adams Abel Ackley Adams Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. Schenectady, NY 12305 Surname Index to Schenectady Births 1940-1953 Adams Adamson Ahl Adams Adanti Ahles Adams Addis Ahman Adams Ademec or Adamec Ahnert Adams Adinolfi Ahren Adams Adinolfi Ahren Adams Adinolfi Ahrendtsen Adams Adinolfi Ahrendtsen Adams Adkins Ahrens Adams Adkins Ahrens Adams Adriance Ahrens Adams Adsit Aiken Adams Aeree Aiken Adams Aernecke Ailes = ? Adams Agans Ainsworth Adams Agans Aker (or Aeher = ?) Adams Aganz (Agans ?) Akers Adams Agare or Abare = ? Akerson Adams Agat Akin Adams Agat Akins Adams Agen Akins Adams Aggen Akland Adams Aggen Albanese Adams Aggen Alberding Adams Aggen Albert Adams Agnew Albert Adams Agnew Albert or Alberti Adams Agnew Alberti Adams Agostara Alberti Adams Agostara (not Agostra) Alberts Adamski Agree Albig Adamski Ahave ? = totally Albig Adamson unclear Albohm Adamson Ahern Albohm Adamson Ahl Albohm (not Albolm) Adamson Ahl Albrezzi Grems-Doolittle Library Schenectady County Historical Society 32 Washington Ave. -
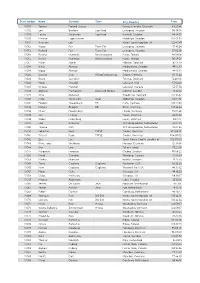
Start Number Name Surname Team City, Country Time 11001 Tommy
Start number Name Surname Team City, Country Time 11001 Tommy Frølund Jensen Kongens lyngby, Denmark 89:20:46 11002 Emil Boström Lag Nord Linköping, Sweden 98:19:04 11003 Lovisa Johansson Lag Nord Burträsk, Sweden 98:19:02 11005 Maurice Lagershausen Huddinge, Sweden 102:01:54 11011 Hyun jun Lee Korea, (south) republic of 122:40:49 11012 Viggo Fält Team Fält Linköping, Sweden 57:45:24 11013 Richard Fält Team Fält Linköping, Sweden 57:45:28 11014 Rasmus Nummela Rackmannarna Pjelax, Finland 98:59:06 11015 Rafael Nummela Rackmannarna Pjelax, Finland 98:59:06 11017 Marie Zølner Hillerød, Denmark 82:11:49 11018 Niklas Åkesson Mellbystrand, Sweden 99:45:22 11019 Viggo Åkesson Mellbystrand, Sweden 99:45:22 11020 Damian Sulik #StopComplaining Siegen, Germany 101:13:24 11023 Bruno Svendsen Tylstrup, Denmark 74:48:08 11024 Philip Oswald Lakeland, USA 55:52:02 11025 Virginia Marshall Lakefield, Canada 55:57:54 11026 Andreas Mathiasson Backyard Heroes Ödsmål, Sweden 75:12:02 11028 Stine Andersen Brædstrup, Denmark 104:35:37 11030 Patte Johansson Tiger Södertälje, Sweden 80:24:51 11031 Frederic Wiesenbach BB Dahn, Germany 101:21:50 11032 Romea Brugger BB Berlin, Germany 101:22:44 11034 Oliver Freudenberg Heyda, Germany 75:07:48 11035 Jan Hennig Farum, Denmark 82:11:50 11036 Robert Falkenberg Farum, Denmark 82:11:51 11037 Edo Boorsma Sint annaparochie, Netherlands 123:17:34 11038 Trijneke Stuit Sint annaparochie, Netherlands 123:17:35 11040 Sebastian Keck TATSE Gieäen, Germany 104:08:01 11041 Tatjana Kage TATSE Gieäen, Germany 104:08:02 11042 Eun Lee -
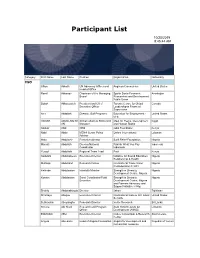
Participant List
Participant List 10/20/2019 8:45:44 AM Category First Name Last Name Position Organization Nationality CSO Jillian Abballe UN Advocacy Officer and Anglican Communion United States Head of Office Ramil Abbasov Chariman of the Managing Spektr Socio-Economic Azerbaijan Board Researches and Development Public Union Babak Abbaszadeh President and Chief Toronto Centre for Global Canada Executive Officer Leadership in Financial Supervision Amr Abdallah Director, Gulf Programs Educaiton for Employment - United States EFE HAGAR ABDELRAHM African affairs & SDGs Unit Maat for Peace, Development Egypt AN Manager and Human Rights Abukar Abdi CEO Juba Foundation Kenya Nabil Abdo MENA Senior Policy Oxfam International Lebanon Advisor Mala Abdulaziz Executive director Swift Relief Foundation Nigeria Maryati Abdullah Director/National Publish What You Pay Indonesia Coordinator Indonesia Yussuf Abdullahi Regional Team Lead Pact Kenya Abdulahi Abdulraheem Executive Director Initiative for Sound Education Nigeria Relationship & Health Muttaqa Abdulra'uf Research Fellow International Trade Union Nigeria Confederation (ITUC) Kehinde Abdulsalam Interfaith Minister Strength in Diversity Nigeria Development Centre, Nigeria Kassim Abdulsalam Zonal Coordinator/Field Strength in Diversity Nigeria Executive Development Centre, Nigeria and Farmers Advocacy and Support Initiative in Nig Shahlo Abdunabizoda Director Jahon Tajikistan Shontaye Abegaz Executive Director International Insitute for Human United States Security Subhashini Abeysinghe Research Director Verite -

Human Genetics and Clinical Aspects of Neurodevelopmental Disorders
bioRxiv preprint doi: https://doi.org/10.1101/000687; this version posted October 6, 2014. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Human genetics and clinical aspects of neurodevelopmental disorders Gholson J. Lyon1,2,3,*, Jason O’Rawe1,3 1Stanley Institute for Cognitive Genomics, One Bungtown Road, Cold Spring Harbor Laboratory, NY, USA, 11724 2Institute for Genomic Medicine, Utah Foundation for Biomedical Research, E 3300 S, Salt Lake City, Salt Lake City, UT, USA, 84106 3Stony Brook University, 100 Nicolls Rd, Stony Brook, NY, USA, 11794 * Corresponding author: Gholson J. Lyon Email: [email protected] Other author emails: Jason O'Rawe: [email protected] 1 bioRxiv preprint doi: https://doi.org/10.1101/000687; this version posted October 6, 2014. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Introduction “our incomplete studies do not permit actual classification; but it is better to leave things by themselves rather than to force them into classes which have their foundation only on paper” – Edouard Seguin (Seguin, 1866) “The fundamental mistake which vitiates all work based upon Mendel’s method is the neglect of ancestry, and the attempt to regard the whole effect upon off- spring, produced by a particular parent, as due to the existence in the parent of particular structural characters; while the contradictory results obtained by those who have observed the offspring of parents apparently identical in cer- tain characters show clearly enough that not only the parents themselves, but their race, that is their ancestry, must be taken into account before the result of pairing them can be predicted” – Walter Frank Raphael Weldon (Weldon, 1902).