Quantum and Quantum-Classical Calculations of Core-Ionized Molecules in Varied Environments
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Lecture #2: August 25, 2020 Goal Is to Define Electrons in Atoms
Lecture #2: August 25, 2020 Goal is to define electrons in atoms • Bohr Atom and Principal Energy Levels from “orbits”; Balance of electrostatic attraction and centripetal force: classical mechanics • Inability to account for emission lines => particle/wave description of atom and application of wave mechanics • Solutions of Schrodinger’s equation, Hψ = Eψ Required boundaries => quantum numbers (and the Pauli Exclusion Principle) • Electron configurations. C: 1s2 2s2 2p2 or [He]2s2 2p2 Na: 1s2 2s2 2p6 3s1 or [Ne] 3s1 => Na+: [Ne] Cl: 1s2 2s2 2p6 3s23p5 or [Ne]3s23p5 => Cl-: [Ne]3s23p6 or [Ar] What you already know: Quantum Numbers: n, l, ml , ms n is the principal quantum number, indicates the size of the orbital, has all positive integer values of 1 to ∞(infinity) (Bohr’s discrete orbits) l (angular momentum) orbital 0s l is the angular momentum quantum number, 1p represents the shape of the orbital, has integer values of (n – 1) to 0 2d 3f ml is the magnetic quantum number, represents the spatial direction of the orbital, can have integer values of -l to 0 to l Other terms: electron configuration, noble gas configuration, valence shell ms is the spin quantum number, has little physical meaning, can have values of either +1/2 or -1/2 Pauli Exclusion principle: no two electrons can have all four of the same quantum numbers in the same atom (Every electron has a unique set.) Hund’s Rule: when electrons are placed in a set of degenerate orbitals, the ground state has as many electrons as possible in different orbitals, and with parallel spin. -

The Quantum Mechanical Model of the Atom
The Quantum Mechanical Model of the Atom Quantum Numbers In order to describe the probable location of electrons, they are assigned four numbers called quantum numbers. The quantum numbers of an electron are kind of like the electron’s “address”. No two electrons can be described by the exact same four quantum numbers. This is called The Pauli Exclusion Principle. • Principle quantum number: The principle quantum number describes which orbit the electron is in and therefore how much energy the electron has. - it is symbolized by the letter n. - positive whole numbers are assigned (not including 0): n=1, n=2, n=3 , etc - the higher the number, the further the orbit from the nucleus - the higher the number, the more energy the electron has (this is sort of like Bohr’s energy levels) - the orbits (energy levels) are also called shells • Angular momentum (azimuthal) quantum number: The azimuthal quantum number describes the sublevels (subshells) that occur in each of the levels (shells) described above. - it is symbolized by the letter l - positive whole number values including 0 are assigned: l = 0, l = 1, l = 2, etc. - each number represents the shape of a subshell: l = 0, represents an s subshell l = 1, represents a p subshell l = 2, represents a d subshell l = 3, represents an f subshell - the higher the number, the more complex the shape of the subshell. The picture below shows the shape of the s and p subshells: (notice the electron clouds) • Magnetic quantum number: All of the subshells described above (except s) have more than one orientation. -

Quantum Spin Systems and Their Local and Long-Time Properties Carolee Wheeler Faculty Advisor: Robert Sims
URA Project Proposal Quantum Spin Systems and Their Local and Long-Time Properties Carolee Wheeler Faculty Advisor: Robert Sims In quantum mechanics, spin is an important concept having to do with atomic nuclei, hadrons, and elementary particles. Spin may be thought of as a measure of a particle’s rotation about its axis. However, spin differs from orbital angular momentum in the sense that the particles may carry integer or half-integer quantum numbers, i.e. 0, 1/2, 1, 3/2, 2, etc., whereas orbital angular momentum may only take integer quantum numbers. Furthermore, the spin of a charged elementary particle is related to a magnetic dipole moment. All quantum mechanic particles have an inherent spin. This is due to the fact that elementary particles (such as photons, electrons, or quarks) cannot be divided into smaller entities. In other words, they cannot be viewed as particles that are made up of individual, smaller particles that rotate around a common center. Thus, the spin that elementary particles carry is an intrinsic property [1]. An important characteristic of spin in quantum mechanics is that it is quantized. The magnitude of spin takes values S = h s(s + )1 , with h being the reduced Planck’s constant and s being the spin quantum number (a non- negative integer or half-integer). Spin may also be viewed in composite particles, and is calculated by summing the spins of the constituent particles. In the case of atoms and molecules, spin is the sum of the spins of unpaired electrons [1]. Particles with spin can possess a magnetic moment. -

The Principal Quantum Number the Azimuthal Quantum Number The
To completely describe an electron in an atom, four quantum numbers are needed: energy (n), angular momentum (ℓ), magnetic moment (mℓ), and spin (ms). The Principal Quantum Number This quantum number describes the electron shell or energy level of an atom. The value of n ranges from 1 to the shell containing the outermost electron of that atom. For example, in caesium (Cs), the outermost valence electron is in the shell with energy level 6, so an electron incaesium can have an n value from 1 to 6. For particles in a time-independent potential, as per the Schrödinger equation, it also labels the nth eigen value of Hamiltonian (H). This number has a dependence only on the distance between the electron and the nucleus (i.e. the radial coordinate r). The average distance increases with n, thus quantum states with different principal quantum numbers are said to belong to different shells. The Azimuthal Quantum Number The angular or orbital quantum number, describes the sub-shell and gives the magnitude of the orbital angular momentum through the relation. ℓ = 0 is called an s orbital, ℓ = 1 a p orbital, ℓ = 2 a d orbital, and ℓ = 3 an f orbital. The value of ℓ ranges from 0 to n − 1 because the first p orbital (ℓ = 1) appears in the second electron shell (n = 2), the first d orbital (ℓ = 2) appears in the third shell (n = 3), and so on. This quantum number specifies the shape of an atomic orbital and strongly influences chemical bonds and bond angles. -

Chem 1A Chapter 7 Exercises
Chem 1A Chapter 7 Exercises Exercises #1 Characteristics of Waves 1. Consider a wave with a wavelength = 2.5 cm traveling at a speed of 15 m/s. (a) How long does it take for the wave to travel a distance of one wavelength? (b) How many wavelengths (or cycles) are completed in 1 second? (The number of waves or cycles completed per second is referred to as the wave frequency.) (c) If = 3.0 cm but the speed remains the same, how many wavelengths (or cycles) are completed in 1 second? (d) If = 1.5 cm and the speed remains the same, how many wavelengths (or cycles) are completed in 1 second? (e) What can you conclude regarding the relationship between wavelength and number of cycles traveled in one second, which is also known as frequency ()? Answer: (a) 1.7 x 10–3 s; (b) 600/s; (c) 500/s; (d) 1000/s; (e) ?) 2. Consider a red light with = 750 nm and a green light = 550 nm (a) Which light travels a greater distance in 1 second, red light or green light? Explain. (b) Which light (red or green) has the greater frequency (that is, contains more waves completed in 1 second)? Explain. (c) What are the frequencies of the red and green light, respectively? (d) If light energy is defined by the quantum formula, such that Ep = h, which light carries more energy per photon? Explain. 8 -34 (e) Assuming that the speed of light, c = 2.9979 x 10 m/s, Planck’s constant, h = 6.626 x 10 J.s., and 23 Avogadro’s number, NA = 6.022 x 10 /mol, calculate the energy: (i) per photon; (ii) per mole of photon for green and red light, respectively. -
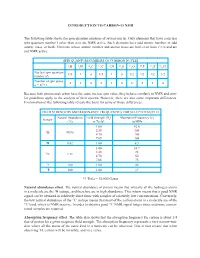
INTRODUCTION to CARBON-13 NMR the Following Table Shows The
INTRODUCTION TO CARBON-13 NMR The following table shows the spin quantum numbers of several nuclei. Only elements that have a nuclear spin quantum number I other than zero are NMR active. Such elements have odd atomic number, or odd atomic mass, or both. Elements whose atomic number and atomic mass are both even have I = 0 and are not NMR active. SPIN QUANTUM NUMBERS OF COMMON NUCLEI 1 2 12 13 14 16 17 19 31 35 1H 1H 6C 6C 7N 8O 8O 9F 15P 17Cl Nuclear spin quantum 1/2 1 0 1/2 1 0 5/2 1/2 1/2 3/2 number (I) Number of spin states 2 3 0 2 3 0 6 2 2 4 n = 2I + 1 Because both proton and carbon have the same nuclear spin value, they behave similarly in NMR and simi- lar guidelines apply to the analysis of their spectra. However, there are also some important differences. Examination of the following table reveals the basis for some of those differences. FIELD STRENGTHS AND RESONANCE FREQUENCIES FOR SELECTED NUCLEI Natural Abundance Field Strength (B ) Absorption Frequency (n) Isotope o (%) in Tesla* in MHz 1.00 42.6 2.35 100 1H 99.98 4.70 200 7.05 300 2H 0.02 1.00 6.5 1.00 10.7 2.35 25 13C 1.11 4.70 50 7.05 75 19F 100 1.00 40 31P 100 1.00 17 *1 Tesla = 10,000 Gauss Natural abundance effect. The natural abundance of proton means that virtually all the hydrogen atoms in a molecule are the 1H isotope, and therefore are in high abundance. -

Free Electron Fermi Gas
Phys463.nb 33 6 Free Electron Fermi Gas 6.1. Electrons in a metal 6.1.1. Electrons in one atom One electron in an atom (a hydrogen-like atom): the nucleon has charge +Z e, where Z is the atomic number, and there is one electron moving around this nucleon Four quantum number: n, l and lz, sz. Energy levels En with n = 1, 2, 3 ... Z2 m e4 1 En = - (6.1) 2 2 2 2 32 p e0 Ñ n where m = me mN me + mN » me where me is the mass of an electron and mN is the mass of the nucleon. 13.6 eV Z2 En = - H L (6.2) n2 H L For each n, the angular momentum quantum number [L2 y = l l + 1 y] can take the values of l = 0, 1, 2, 3, 4 … , n - 1. These states are known as the s, p, d, f , g,… states For each l, the quantum number for Lz can be any integer betweenH -Ll and +l lz = -l, -l + 1,…, l - 1, l . For fixed n, l and lz, the spin quantum number sz can be +1/2 or -1/2 (up or down). H L At each n, there are 2 n2 quantum states. In a real atom In a hydrogen-like atom, for the same n, all the 2 n2 quantum number has the same energy. In a real atom, the energy level splits according to the total angular momentum quantum number l. Typically, the state with lower l has lower energy (not always true). -

Part 1. the Electronic Structure of Atoms Hydrogen-Like Atoms
Part 1. The Electronic Structure of Atoms The wave function of an atom contains all the information about the properties of the atom. The wave functions for the hydrogen atom and other one-electron atoms (such as He+ and Li2+) can be calculated exactly by solving Schrödinger’s equation. Approximate methods must be used to calculate the wave functions for atoms containing two or more electrons. These numerical calculations can be very accurate, but require complicated computer calculations. So we start with (brief review from Chem 331): Hydrogen-Like Atoms What are they? He+ (Z = 2) , Li2+ (Z = 3), Be3+ (Z = 4), etc. Ions with one nucleus and one electron (two bound particles), atomic number Z > 1. Also a few rare, but interesting, “exotics”, such as muonium (more on that in the tutorial!). Why are they important? Schrödinger’s equation can be solved exactly for hydrogen-like atoms, using the reduced mass concept to simplify the two-body problem to an effective one-body problem. This greatly expands the number of chemical systems that can be treated analytically by quantum mechanical principles. (For molecules and multi-electron atoms, cumbersome and less accurate numerical methods must be used to solve Schrödinger’s equation.) For hydrogen-like atoms, the electrostatic potential energy and the reduced mass in Schrödinger’s equation for the hydrogen atom e2 V (r) = − 40r m m = e p me + mp are changed to (notice the factor of Z) Ze 2 V (r) = − 40r m m = e N me + mN me is the electron mass and mN is the nucleus mass. -

Spin Quantum Number Or Rest Angular Momentum
Spin Quantum Number or Rest Angular Momentum H. Razmi (1) and A. MohammadKazemi (2) Department of Physics, The University of Qom, The Old Isfahan Road, Qom, I. R. Iran. (1) [email protected] & [email protected] (2) [email protected] Abstract The aim of this letter (considering the fundamental origins of Klein-Gordon and Dirac equations, Thomas precession, and the photon spin) is to point out that since the origin of spin angular momentum refers to relativity than quantum theory, it is better to call it as the “rest angular momentum”. 1 Spin quantity which was introduced short after formulation of quantum mechanics, in addition to its theoretical significance, has a wide variety of applications in different technological fields (e. g. NMR and new hot subject of research spintronics). Although the mathematical formulation (Lie algebra) of the subject is well-known, the fundamental discussions related to the spin can be hardly found in modern (text)books and papers. In this letter, with an overview of the fundamental origins of Klein-Gordon and Dirac equations, Thomas precession, and the photon spin, it is pointed out that the origin of spin angular momentum refers to relativity than quantum theory. As is clear in the history of the discovery of the electron spin [1], spin degree of freedom cannot be considered as the electron rotation. If the electron is to rotate around itself, its surface velocity should be more than the velocity of light in vacuum?! Although some experts have tried to find a realistic and causal model for spin angular momentum [2], they haven't succeeded to satisfy the physics society yet; the reason is that the so-called classical models are "toy" models than be considered realistic. -

Lecture 3 2014 Lecture.Pdf
Chapter 2 ATOMIC STRUCTURE AND INTERATOMIC BONDING Chapter 2: Main Concepts 1. History of atomic models: from ancient Greece to Quantum mechanics 2. Quantum numbers 3. Electron configurations of elements 4. The Periodic Table 5. Bonding Force and Energies 6. Electron structure and types of atomic bonds 7. Additional: How to see atoms: Transmission Electron Microscopy Topics 1, 2 and partially 3: Lecture 3 Topic 5: Lecture 4 Topic 7: Lecture 5 Topics 4&6: self-education (book and/or WileyPlus) Question 1: What are the different levels of Material Structure? • Atomic structure (~1Angstrem=10-10 m) • Crystalline structure (short and long-range atomic arrangements; 1-10Angstroms) • Nanostructure (1-100nm) • Microstructure (0.1 -1000 mm) • Macrostructure (>1000 mm) Q2: How does atomic structure influence the Materials Properties? • In general atomic structure defines the type of bonding between elements • In turn the bonding type (ionic, metallic, covalent, van der Waals) influences the variety of materials properties (module of elasticity, electro and thermal conductivity and etc.). MATERIALS CLASSIFICATION • For example, five groups of materials can be outlined based on structures and properties: - metals and alloys - ceramics and glasses - polymers (plastics) - semiconductors - composites Historical Overview A-tomos On philosophical grounds: There must be a smallest indivisible particle. Arrangement of different Democritus particles at micro-scale determine properties at 460-~370 BC macro-scale. It started with … fire dry hot air earth Aristoteles wet cold 384-322 BC water The four elements Founder of Logic and from ancient times Methodology as tools for Science and Philosophy Science? Newton published in 1687: Newton ! ‘Philosphiae Naturalis Principia Mathematica’, (1643-1727) … while the alchemists were still in the ‘dark ages’. -

Quantum Numbers
3.091: Introduction to Solid State Chemistry Maddie Sutula, Fall 2018 Recitation 5 1 Quantum numbers Every electron localized in an atom can be described by four quantum numbers. The Pauli Exclusion Principle tells us that no two electrons can share the exact same set of quantum numbers Principal quantum number The principle quantum number, n, represents the energy level of the electron, much like the n used in the Bohr model. Energy is related to the principle quantum number by 13:6 eV E = − ; n > 0 n2 Orbital angular momentum quantum number The orbital angular momentum quantum number, l, provides information about the shape of an orbital. Unlike the description of early models of the atom, electrons in atoms don't orbit around the nucleus like a planet around the sun. However, they do have angular momentum, and the shape of the probability cloud around the nucleus depends on the value of the angular momentum. The magnitude of the angular momentum is related to the orbital angular momentum quantum number by p L = ¯h l(l + 1); 0 ≤ l ≤ n − 1 The orbital angular momentum quantum numbers correspond directly to letters commonly used by chemists to describe the orbital subshells: for example, l = 1 corresponds to the s orbitals, which are spherical: l = 2 corresponds to the p orbitals, which are lobed: Visualizations of the higher orbitals, like the d (l − 2) and f (l = 3) can be found in Chapter 6 of the Averill textbook. Images of orbitals from B. A. Averill and P. Eldredge, General Chemistry. License: CC BY-NC-SA. -

Electron Configurations
Electron Configurations The location of electrons in orbit around the nucleus is what determines how an element will behave in chemical reactions. Therefore; electron configuration is everything to chemistry. There are three ways to show electron configuration: Orbital Notation Electron Configuration Notation Lewis Dot Notation Quantum Mechanical Model of the Atom (today’s model) Electrons DO occupy energy levels but their path is not definite Energy Increases You cannot say with certainty away from the that an electron will be at any nucleus point at any given time, although you do know that there is a HIGH PROBABILITY that the electrons are around the nucleus Energy Levels – regions around the nucleus where electrons are likely to be found Remember the ladder…each step is like an energy level Quantum Numbers Quantum Numbers – numbers that describe atomic orbitals and their electrons Orbitals – the place within an energy level where an electron will probably be found There are 4 quantum numbers! (1) PRINCIPAL Quantum Number – n describes the principal (main) energy levels of electrons in an atom the main energy levels are known as SHELLS increase in n = increase in energy maximum number of electrons (e-) in an energy level = 2n (2) ORBITAL Quantum Number – l determines the energy sublevel AND the shape of the orbital the number of sublevels is equal to the principal quantum number four shapes of the energy sub levels are: s = spherical p = dumbbell d = double dumbbell f = too complex to describe P Orbital S Orbital