Medications for Duchenne Muscular Dystrophy SERVICE: Casimersen
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

DRUGS REQUIRING PRIOR AUTHORIZATION in the MEDICAL BENEFIT Page 1
Effective Date: 08/01/2021 DRUGS REQUIRING PRIOR AUTHORIZATION IN THE MEDICAL BENEFIT Page 1 Therapeutic Category Drug Class Trade Name Generic Name HCPCS Procedure Code HCPCS Procedure Code Description Anti-infectives Antiretrovirals, HIV CABENUVA cabotegravir-rilpivirine C9077 Injection, cabotegravir and rilpivirine, 2mg/3mg Antithrombotic Agents von Willebrand Factor-Directed Antibody CABLIVI caplacizumab-yhdp C9047 Injection, caplacizumab-yhdp, 1 mg Cardiology Antilipemic EVKEEZA evinacumab-dgnb C9079 Injection, evinacumab-dgnb, 5 mg Cardiology Hemostatic Agent BERINERT c1 esterase J0597 Injection, C1 esterase inhibitor (human), Berinert, 10 units Cardiology Hemostatic Agent CINRYZE c1 esterase J0598 Injection, C1 esterase inhibitor (human), Cinryze, 10 units Cardiology Hemostatic Agent FIRAZYR icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent HAEGARDA c1 esterase J0599 Injection, C1 esterase inhibitor (human), (Haegarda), 10 units Cardiology Hemostatic Agent ICATIBANT (generic) icatibant J1744 Injection, icatibant, 1 mg Cardiology Hemostatic Agent KALBITOR ecallantide J1290 Injection, ecallantide, 1 mg Cardiology Hemostatic Agent RUCONEST c1 esterase J0596 Injection, C1 esterase inhibitor (recombinant), Ruconest, 10 units Injection, lanadelumab-flyo, 1 mg (code may be used for Medicare when drug administered under Cardiology Hemostatic Agent TAKHZYRO lanadelumab-flyo J0593 direct supervision of a physician, not for use when drug is self-administered) Cardiology Pulmonary Arterial Hypertension EPOPROSTENOL (generic) -

Opportunities and Challenges for Antisense Oligonucleotide Therapies
Received: 10 January 2020 Revised: 23 April 2020 Accepted: 8 May 2020 DOI: 10.1002/jimd.12251 REVIEW ARTICLE Opportunities and challenges for antisense oligonucleotide therapies Elsa C. Kuijper1 | Atze J. Bergsma2,3 | W.W.M. Pim Pijnappel2,3 | Annemieke Aartsma-Rus1 1Department of Human Genetics, Leiden University Medical Center, Leiden, The Abstract Netherlands Antisense oligonucleotide (AON) therapies involve short strands of modi- 2Department of Pediatrics, Center for fied nucleotides that target RNA in a sequence-specific manner, inducing Lysosomal and Metabolic Diseases, targeted protein knockdown or restoration. Currently, 10 AON therapies Erasmus Medical Center, Rotterdam, The Netherlands have been approved in the United States and Europe. Nucleotides are chem- 3Department of Clinical Genetics, Center ically modified to protect AONs from degradation, enhance bioavailability for Lysosomal and Metabolic Diseases, and increase RNA affinity. Whereas single stranded AONs can efficiently Erasmus Medical Center, Rotterdam, The Netherlands be delivered systemically, delivery of double stranded AONs requires capsulation in lipid nanoparticles or binding to a conjugate as the uptake Correspondence enhancing backbone is hidden in this conformation. With improved chem- Annemieke Aartsma-Rus, LUMC Postzone S4-P, Albinusdreef 2, 2333 ZA istry, delivery vehicles and conjugates, doses can be lowered, thereby reduc- Leiden, The Netherlands. ing the risk and occurrence of side effects. AONs can be used to knockdown Email: [email protected] or restore levels of protein. Knockdown can be achieved by single stranded Communicating Editor: Carla E. Hollak or double stranded AONs binding the RNA transcript and activating RNaseH-mediated and RISC-mediated degradation respectively. Transcript binding by AONs can also prevent translation, hence reducing protein levels. -

Duchenne Muscular Dystrophy (DMD) Agents
Therapeutic Class Overview Duchenne muscular dystrophy (DMD) Agents INTRODUCTION • Duchenne muscular dystrophy (DMD) is 1 of 4 conditions known as dystrophinopathies, which are inherited, X-linked myopathic disorders due to a defect in the dystrophin gene that results in the primary pathologic process of muscle fiber degradation. The hallmark symptom is progressive weakness (Darras 2018[a], Darras 2018[b], Muscular Dystrophy Association [MDA] 2019). The other 3 conditions include: Becker muscular dystrophy (BMD), which is a mild form of DMD; an intermediate presentation between BMD and DMD; and DMD-associated dilated cardiomyopathy, which has little or no clinical ○ skeletal or muscle disease (MDA 2019). • DMD symptom onset is in early childhood, usually between the ages of 2 and 3 years old. The proximal muscles are affected first, followed by the distal limb muscles. Generally, the lower external muscles will be affected before the upper. The affected child may have difficulties jumping, walking, and running (MDA 2019). • The prevalence of DMD ranges from 1 to 2 per 10,000 live male births; female-manifesting carriers are rarer, but can present with a range of symptoms that vary in their severities (Birnkrant et al 2018, Darras 2018[a], Emflaza Food and Drug Administration [FDA] Medical Review 2017). • The clinical course and lifespan of patients with DMD is relatively short. Individuals are usually confined to a wheelchair by age 13, and many die in their late teens or twenties from respiratory insufficiency or cardiomyopathy. Although survival until adulthood is more common now, very few patients survive past the 3rd decade (Darras 2018[a]). -

Vyondys 53™ (Golodirsen)
UnitedHealthcare® Commercial Medical Benefit Drug Policy Vyondys 53™ (Golodirsen) Policy Number: 2021D0088C Effective Date: April 1, 2021 Instructions for Use Table of Contents Page Related Commercial Policy Coverage Rationale ....................................................................... 1 • Provider Administered Drugs – Site of Care Applicable Codes .......................................................................... 2 Background.................................................................................... 2 Community Plan Policy Benefit Considerations .................................................................. 3 • Vyondys 53™ (Golodirsen) Clinical Evidence ........................................................................... 3 U.S. Food and Drug Administration ............................................. 3 References ..................................................................................... 4 Policy History/Revision Information ............................................. 4 Instructions for Use ....................................................................... 4 Coverage Rationale See Benefit Considerations Vyondys 53 (golodirsen) may be covered for the treatment of Duchenne muscular dystrophy (DMD) in patients who meet all of the following criteria: For initial therapy, all of the following: o Diagnosis of Duchenne muscular dystrophy by, or in consultation with, a neurologist with expertise in the diagnosis of DMD; and o Submission of medical records (e.g., chart notes, laboratory -

Prescription Drugs Requiring Prior Authorization
PRESCRIPTION DRUGS REQUIRING PRIOR AUTHORIZATION Revised 10/16 As part of our drug utilization management program, members must request and receive prior authorization for certain prescription drugs in order to use their prescription drug benefits. Below is a list of drugs that currently require prior authorization. This list will be updated periodically as new drugs that require prior authorization are introduced. As benefits may vary by group and individual plans, the inclusion of a medication on this list does not imply prescription drug coverage. The Schedule of Benefits contains a list of drug categories that require prior authorization. Prior authorization requests are processed by our pharmacy benefit manager, Express Scripts, Inc. (ESI). Physicians must call ESI to obtain an authorization. (1-800-842-2015). Drug Name Generic Name Drug Classification Abstral fentanyl citrate oral tablet Controlled Dangerous Substance Accu-Chek Test Strips blood glucose test strips Blood Glucose Test Strips Actemra tocilizumab Monoclonal Antibody Acthar corticotropin Hormone Actimmune interferon gamma 1b Interferon Actiq fentanyl citrate OTFC Controlled Dangerous Substance Adcirca tadalafil Pulmonary Vasodilator Adempas riociguat Pulmonary Vasodilator Adlyxin lixisenatide Type 2 Diabetes Advocate Test Strips blood glucose test strips Blood Glucose Test Strips Aerospan** flunisolide Corticosteroids (Inhaled) Afrezza insulin Insulin (inhaled) Ampyra dalfampridine Multiple Sclerosis Agent Altoprev** lovastatin Cholesterol Alvesco** ciclesonide Corticosteroids -

The Use of Ataluren in the Effective Management of Duchenne Muscular Dystrophy
Review Neuromuscular Diseases Early Diagnosis and Treatment – The Use of Ataluren in the Effective Management of Duchenne Muscular Dystrophy Eugenio Mercuri,1 Ros Quinlivan2 and Sylvie Tuffery-Giraud3 1. Catholic University, Rome, Italy; 2. Great Ormond Street Hospital and National Hospital for Neurology and Neurosurgery, London, UK; 3. Laboratory of Genetics of Rare Diseases (LGMR), University of Montpellier, Montpellier, France DOI: https://doi.org/10.17925/ENR.2018.13.1.31 he understanding of the natural history of Duchenne muscular dystrophy (DMD) is increasing rapidly and new treatments are emerging that have the potential to substantially improve the prognosis for patients with this disabling and life-shortening disease. For many, Thowever, there is a long delay between the appearance of symptoms and DMD diagnosis, which reduces the possibility of successful treatment. DMD results from mutations in the large dystrophin gene of which one-third are de novo mutations and two-thirds are inherited from a female carrier. Roughly 75% of mutations are large rearrangements and 25% are point mutations. Certain deletions and nonsense mutations can be treated whereas many other mutations cannot currently be treated. This emphasises the need for early genetic testing to identify the mutation, guide treatment and inform genetic counselling. Treatments for DMD include corticosteroids and more recently, ataluren has been approved in Europe, the first disease-modifying therapy for treating DMD caused by nonsense mutations. The use of ataluren in DMD is supported by positive results from phase IIb and phase III studies in which the treatment produced marked improvements in the 6-minute walk test, timed function tests such as the 10 m walk/run test and the 4-stair ascent/descent test compared with placebo. -

Refreshing the Biologic Pipeline 2020
news feature Credit: Science Lab / Alamy Stock Photo Refreshing the biologic pipeline 2020 In the absence of face-to-face meetings, FDA and industry implemented regulatory workarounds to maintain drug and biologics approvals. These could be here to stay. John Hodgson OVID-19 might have been expected since 1996) — a small miracle in itself “COVID-19 confronted us with the need to severely impair drug approvals (Fig. 1 and Table 1). to better triage sponsors’ questions,” says Cin 2020. In the event, however, To the usual crop of rare disease and Peter Marks, the director of the Center for industry and regulators delivered a small genetic-niche cancer treatments, 2020 Biologics Evaluation and Research (CBER) miracle. They found workarounds and also added a chimeric antigen receptor at the FDA. “That was perhaps the single surrogate methods of engagement. Starting (CAR)-T cell therapy with a cleaner biggest takeaway from the pandemic related in January 2020, when the outbreak veered manufacturing process and the first to product applications.” Marks says that it westward, the number of face-to face approved blockbuster indication for a became very apparent with some COVID- meetings declined rapidly; by March, small-interfering RNA (siRNA) — the 19-related files that resolving a single they were replaced by Webex and Teams. European Medicines Agency’s (EMA) issue can help a sponsor enormously and (Secure Zoom meeting are to be added registration of the RNA interference accelerate the development cycle. Before this year.) And remarkably, by 31 December, (RNAi) therapy Leqvio (inclisiran) for COVID-19, it was conceivable that a small the US Food and Drug Administration cardiovascular disease. -

Increased Dystrophin Production with Golodirsen in Patients with Duchenne Muscular Dystrophy
Published Ahead of Print on March 5, 2020 as 10.1212/WNL.0000000000009233 ARTICLE OPEN ACCESS CLASS OF EVIDENCE Increased dystrophin production with golodirsen in patients with Duchenne muscular dystrophy Diane E. Frank, PhD, Frederick J. Schnell, PhD, Cody Akana, BS, Saleh H. El-Husayni, BS, Correspondence Cody A. Desjardins, PhD, Jennifer Morgan, PhD, Jay S. Charleston, PhD, Valentina Sardone, PhD, Dr. Muntoni Joana Domingos, MD, George Dickson, PhD, Volker Straub, MD, Michela Guglieri, Eugenio Mercuri, MD, [email protected] Laurent Servais, PhD, and Francesco Muntoni, MD, on behalf of the SKIP-NMD Study Group Neurology® 2020;00:1-13. doi:10.1212/WNL.0000000000009233 Abstract MORE ONLINE Objective Class of Evidence To report safety, pharmacokinetics, exon 53 skipping, and dystrophin expression in golodirsen- Criteria for rating treated patients with Duchenne muscular dystrophy (DMD) amenable to exon 53 skipping. therapeutic and diagnostic studies Methods NPub.org/coe Part 1 was a randomized, double-blind, placebo-controlled, 12-week dose titration of once-weekly golodirsen; part 2 is an ongoing, open-label evaluation. Safety and pharmacokinetics were primary and secondary objectives of part 1. Primary biological outcome measures of part 2 were blinded exon skipping and dystrophin protein production on muscle biopsies (baseline, week 48) evaluated, respectively, using reverse transcription PCR and Western blot and immunohistochemistry. Results Twelve patients were randomized to receive golodirsen (n = 8) or placebo (n = 4) in part 1. All from part 1 plus 13 additional patients received 30 mg/kg golodirsen in part 2. Safety findings were consistent with those previously observed in pediatric patients with DMD. -

Animal Models of Duchenne Muscular Dystrophy: from Basic Mechanisms to Gene Therapy Joe W
© 2015. Published by The Company of Biologists Ltd | Disease Models & Mechanisms (2015) 8, 195-213 doi:10.1242/dmm.018424 REVIEW Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy Joe W. McGreevy1, Chady H. Hakim1, Mark A. McIntosh1 and Dongsheng Duan1,2,* ABSTRACT The identification of the disease-causing gene and the molecular Duchenne muscular dystrophy (DMD) is a progressive muscle- basis for the DMD and BMD phenotypes establishes the foundation wasting disorder. It is caused by loss-of-function mutations in the for DMD gene therapy (Fig. 2A). To mitigate muscle disease, one dystrophin gene. Currently, there is no cure. A highly promising can either restore the full-length transcript or express a truncated but therapeutic strategy is to replace or repair the defective dystrophin in-frame dystrophin gene (Duan, 2011; Goyenvalle et al., 2011; gene by gene therapy. Numerous animal models of DMD have been Konieczny et al., 2013; Mendell et al., 2012; Verhaart and Aartsma- developed over the last 30 years, ranging from invertebrate to large Rus, 2012). Several gene therapy strategies are currently under mammalian models. mdx mice are the most commonly employed development. They include replacing the mutated gene with a models in DMD research and have been used to lay the groundwork functional candidate gene (gene replacement) or repairing the for DMD gene therapy. After ~30 years of development, the field has defective gene by targeted correction and exon skipping (gene reached the stage at which the results in mdx mice can be validated repair). Currently, adeno-associated virus (AAV)-mediated gene and scaled-up in symptomatic large animals. -
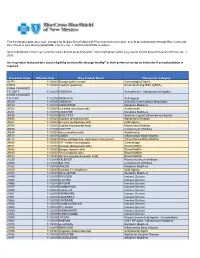
Procedure Code Effective Date Drug Product Name* Therapeutic
This list includes procedure code changes for Medical Benefit Specialty Pharmacy that may require benefit preauthorization through Blue Cross and Blue Shield of New Mexico (BCBSNM) effective Jan. 1, 2020 for BCBSNM members. Items highlighted in blue may currently require benefit preauthorization. Items highlighted yellow may require benefit preauthorization effective Jan. 1, 2020. It is imperative that providers check eligibility and benefits through Availity® or their preferred vendor to determine if preauthorization is required. Procedure Code Effective Date Drug Product Name* Therapeutic Category 90378 1/1/2020 Synagis (palivizumab) Immunological Agent C9036 1/1/2020 Onpattro (patisiran) Small interfering RNA (siRNA) C9466 CHANGED TO J0517 1/1/2019 FASENRA Antiasthmatic - Monoclonal Antibodies C9493 CHANGED TO J1301 1/1/2019 RADICAVA ALS Agents J0129 1/1/2019 ORENCIA Selective Costimulation Modulators J0180 1/1/2019 FABRAZYME Metabolic Modifiers J0202 1/1/2020 Lemtrada (alemtuzumab) Autoimmune J0221 1/1/2019 LUMIZYME Metabolic Modifiers J0490 1/1/2019 BENLYSTA Systemic Lupus Erythematosus Agents J0565 1/1/2020 Zinplava (bezlotoxumab) Monoclonal Antibody J0567 1/1/2020 Brineura (cerliponase alfa) CNS Agents J0584 1/1/2020 Crysvita (burosumab-twza) Monoclonal Antibody J0598 1/1/2019 CINRYZE Complement Inhibitors J0638 1/1/2020 Ilaris (canakinumab) Autoimmune J0717 1/1/2019 CIMZIA Inflammatory Bowel Agents J0775 1/1/2020 Xiaflex (collagenase, clostridium histolyticum) Tissue Permeability Modifier J0800 1/1/2020 H.P. Acthar (corticotropin) -

213026Orig1s000
CENTER FOR DRUG EVALUATION AND RESEARCH APPLICATION NUMBER: 213026Orig1s000 OTHER REVIEW(S) IMMUNOGENICITY ASSESSMENT Application Type NDA Application Number 213026 Submit Date 01/10/2020 Received Date 01/10/2020 Division/Office CDER/OND/ON/DNI Review Completion Date 01/10/2021 Product Name Casimersen Proposed Proprie tary AMONDYS 45 Name Error! Bookmark not defined. Pharmacologic Class PMO exon Skipping Applicant Sarepta Therapeutics, Inc. (b) (4) Applicant Proposed Duchenne muscular dystrophy (DMD) in Indication(s) patients who have a confirmed mutation of the DMD gene that is amenable to exon 45 skipping. Immunogenicity Assessors Primary Assessor(s) Seth Thacker PhD Secondary Assessor (s) Daniela Verthelyi PhD MD Assessor Recommendation: The sponsor has submitted data for anti-dystrophin antibodies in the casimersen trials. These data were generated using assays that were developed for assessing anti-dystrophin antibodies in patients treated with eteplirsen and golodirsen and have already been deemed fit for use. The sponsor submitted anti-dystrophin ADA data for Study 4045-101, which had 12 patients enrolled No positive samples were found. The FPR for these assays in Study 4045-101 were 1.3%(IgG), 7.9% (IgE), and 39% (IgM) as calculated by the assessor. The sponsors has not submitted an assay for the detection of Casimersen-specific ADAs or provided a plan on how they assess the risk associated with the generation of novel epitopes in the dystrophin formed by exon 45 skipping. PMRs will be issued to the sponsor to develop and validate the assays and to assess the patients in study 4045-101 and 4045-301 for Abs to the product and to the peptide generated through the exon skipping strategy. -

Rxoutlook® 1St Quarter 2019
® RxOutlook 1st Quarter 2020 optum.com/optumrx a RxOutlook 1st Quarter 2020 Orphan drugs continue to feature prominently in the drug development pipeline In 1983 the Orphan Drug Act was signed into law. Thirty seven years later, what was initially envisioned as a minor category of drugs has become a major part of the drug development pipeline. The Orphan Drug Act was passed by the United States Congress in 1983 in order to spur drug development for rare conditions with high unmet need. The legislation provided financial incentives to manufacturers if they could demonstrate that the target population for their drug consisted of fewer than 200,000 persons in the United States, or that there was no reasonable expectation that commercial sales would be sufficient to recoup the developmental costs associated with the drug. These “Orphan Drug” approvals have become increasingly common over the last two decades. In 2000, two of the 27 (7%) new drugs approved by the FDA had Orphan Designation, whereas in 2019, 20 of the 48 new drugs (42%) approved by the FDA had Orphan Designation. Since the passage of the Orphan Drug Act, 37 years ago, additional regulations and FDA designations have been implemented in an attempt to further expedite drug development for certain serious and life threatening conditions. Drugs with a Fast Track designation can use Phase 2 clinical trials to support FDA approval. Drugs with Breakthrough Therapy designation can use alternative clinical trial designs instead of the traditional randomized, double-blind, placebo-controlled trial. Additionally, drugs may be approved via the Accelerated Approval pathway using surrogate endpoints in clinical trials rather than clinical outcomes.