Integrating Transcriptomics and Metabolomics for the Analysis of The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Materials: Evaluation of Cytotoxicity and Α-Glucosidase Inhibitory Activity of Amide and Polyamino-Derivatives of Lupane Triterpenoids
Supplementary Materials: Evaluation of cytotoxicity and α-glucosidase inhibitory activity of amide and polyamino-derivatives of lupane triterpenoids Oxana B. Kazakova1*, Gul'nara V. Giniyatullina1, Akhat G. Mustafin1, Denis A. Babkov2, Elena V. Sokolova2, Alexander A. Spasov2* 1Ufa Institute of Chemistry of the Ufa Federal Research Centre of the Russian Academy of Sciences, 71, pr. Oktyabrya, 450054 Ufa, Russian Federation 2Scientific Center for Innovative Drugs, Volgograd State Medical University, Novorossiyskaya st. 39, Volgograd 400087, Russian Federation Correspondence Prof. Dr. Oxana B. Kazakova Ufa Institute of Chemistry of the Ufa Federal Research Centre of the Russian Academy of Sciences 71 Prospeсt Oktyabrya Ufa, 450054 Russian Federation E-mail: [email protected] Prof. Dr. Alexander A. Spasov Scientific Center for Innovative Drugs of the Volgograd State Medical University 39 Novorossiyskaya st. Volgograd, 400087 Russian Federation E-mail: [email protected] Figure S1. 1H and 13C of compound 2. H NH N H O H O H 2 2 Figure S2. 1H and 13C of compound 4. NH2 O H O H CH3 O O H H3C O H 4 3 Figure S3. Anticancer screening data of compound 2 at single dose assay 4 Figure S4. Anticancer screening data of compound 7 at single dose assay 5 Figure S5. Anticancer screening data of compound 8 at single dose assay 6 Figure S6. Anticancer screening data of compound 9 at single dose assay 7 Figure S7. Anticancer screening data of compound 12 at single dose assay 8 Figure S8. Anticancer screening data of compound 13 at single dose assay 9 Figure S9. Anticancer screening data of compound 14 at single dose assay 10 Figure S10. -

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

Allele-Specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish
Allele-specific Expression of Ribosomal Protein Genes in Interspecific Hybrid Catfish by Ailu Chen A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama August 1, 2015 Keywords: catfish, interspecific hybrids, allele-specific expression, ribosomal protein Copyright 2015 by Ailu Chen Approved by Zhanjiang Liu, Chair, Professor, School of Fisheries, Aquaculture and Aquatic Sciences Nannan Liu, Professor, Entomology and Plant Pathology Eric Peatman, Associate Professor, School of Fisheries, Aquaculture and Aquatic Sciences Aaron M. Rashotte, Associate Professor, Biological Sciences Abstract Interspecific hybridization results in a vast reservoir of allelic variations, which may potentially contribute to phenotypical enhancement in the hybrids. Whether the allelic variations are related to the downstream phenotypic differences of interspecific hybrid is still an open question. The recently developed genome-wide allele-specific approaches that harness high- throughput sequencing technology allow direct quantification of allelic variations and gene expression patterns. In this work, I investigated allele-specific expression (ASE) pattern using RNA-Seq datasets generated from interspecific catfish hybrids. The objective of the study is to determine the ASE genes and pathways in which they are involved. Specifically, my study investigated ASE-SNPs, ASE-genes, parent-of-origins of ASE allele and how ASE would possibly contribute to heterosis. My data showed that ASE was operating in the interspecific catfish system. Of the 66,251 and 177,841 SNPs identified from the datasets of the liver and gill, 5,420 (8.2%) and 13,390 (7.5%) SNPs were identified as significant ASE-SNPs, respectively. -

Role of Mitochondrial Ribosomal Protein S18-2 in Cancerogenesis and in Regulation of Stemness and Differentiation
From THE DEPARTMENT OF MICROBIOLOGY TUMOR AND CELL BIOLOGY (MTC) Karolinska Institutet, Stockholm, Sweden ROLE OF MITOCHONDRIAL RIBOSOMAL PROTEIN S18-2 IN CANCEROGENESIS AND IN REGULATION OF STEMNESS AND DIFFERENTIATION Muhammad Mushtaq Stockholm 2017 All previously published papers were reproduced with permission from the publisher. Published by Karolinska Institutet. Printed by E-Print AB 2017 © Muhammad Mushtaq, 2017 ISBN 978-91-7676-697-2 Role of Mitochondrial Ribosomal Protein S18-2 in Cancerogenesis and in Regulation of Stemness and Differentiation THESIS FOR DOCTORAL DEGREE (Ph.D.) By Muhammad Mushtaq Principal Supervisor: Faculty Opponent: Associate Professor Elena Kashuba Professor Pramod Kumar Srivastava Karolinska Institutet University of Connecticut Department of Microbiology Tumor and Cell Center for Immunotherapy of Cancer and Biology (MTC) Infectious Diseases Co-supervisor(s): Examination Board: Professor Sonia Lain Professor Ola Söderberg Karolinska Institutet Uppsala University Department of Microbiology Tumor and Cell Department of Immunology, Genetics and Biology (MTC) Pathology (IGP) Professor George Klein Professor Boris Zhivotovsky Karolinska Institutet Karolinska Institutet Department of Microbiology Tumor and Cell Institute of Environmental Medicine (IMM) Biology (MTC) Professor Lars-Gunnar Larsson Karolinska Institutet Department of Microbiology Tumor and Cell Biology (MTC) Dedicated to my parents ABSTRACT Mitochondria carry their own ribosomes (mitoribosomes) for the translation of mRNA encoded by mitochondrial DNA. The architecture of mitoribosomes is mainly composed of mitochondrial ribosomal proteins (MRPs), which are encoded by nuclear genomic DNA. Emerging experimental evidences reveal that several MRPs are multifunctional and they exhibit important extra-mitochondrial functions, such as involvement in apoptosis, protein biosynthesis and signal transduction. Dysregulations of the MRPs are associated with severe pathological conditions, including cancer. -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -

S41598-018-25035-1.Pdf
www.nature.com/scientificreports OPEN An Innovative Approach for The Integration of Proteomics and Metabolomics Data In Severe Received: 23 October 2017 Accepted: 9 April 2018 Septic Shock Patients Stratifed for Published: xx xx xxxx Mortality Alice Cambiaghi1, Ramón Díaz2, Julia Bauzá Martinez2, Antonia Odena2, Laura Brunelli3, Pietro Caironi4,5, Serge Masson3, Giuseppe Baselli1, Giuseppe Ristagno 3, Luciano Gattinoni6, Eliandre de Oliveira2, Roberta Pastorelli3 & Manuela Ferrario 1 In this work, we examined plasma metabolome, proteome and clinical features in patients with severe septic shock enrolled in the multicenter ALBIOS study. The objective was to identify changes in the levels of metabolites involved in septic shock progression and to integrate this information with the variation occurring in proteins and clinical data. Mass spectrometry-based targeted metabolomics and untargeted proteomics allowed us to quantify absolute metabolites concentration and relative proteins abundance. We computed the ratio D7/D1 to take into account their variation from day 1 (D1) to day 7 (D7) after shock diagnosis. Patients were divided into two groups according to 28-day mortality. Three diferent elastic net logistic regression models were built: one on metabolites only, one on metabolites and proteins and one to integrate metabolomics and proteomics data with clinical parameters. Linear discriminant analysis and Partial least squares Discriminant Analysis were also implemented. All the obtained models correctly classifed the observations in the testing set. By looking at the variable importance (VIP) and the selected features, the integration of metabolomics with proteomics data showed the importance of circulating lipids and coagulation cascade in septic shock progression, thus capturing a further layer of biological information complementary to metabolomics information. -

A Metabolomics Approach to Pharmacotherapy Personalization
Journal of Personalized Medicine Review A Metabolomics Approach to Pharmacotherapy Personalization Elena E. Balashova *, Dmitry L. Maslov and Petr G. Lokhov Institute of Biomedical Chemistry, Pogodinskaya St. 10, Moscow 119121, Russia; [email protected] (D.L.M.); [email protected] (P.G.L.) * Correspondence: [email protected] Received: 29 June 2018; Accepted: 3 September 2018; Published: 5 September 2018 Abstract: The optimization of drug therapy according to the personal characteristics of patients is a perspective direction in modern medicine. One of the possible ways to achieve such personalization is through the application of “omics” technologies, including current, promising metabolomics methods. This review demonstrates that the analysis of pre-dose metabolite biofluid profiles allows clinicians to predict the effectiveness of a selected drug treatment for a given individual. In the review, it is also shown that the monitoring of post-dose metabolite profiles could allow clinicians to evaluate drug efficiency, the reaction of the host to the treatment, and the outcome of the therapy. A comparative description of pharmacotherapy personalization (pharmacogenomics, pharmacoproteomics, and therapeutic drug monitoring) and personalization based on the analysis of metabolite profiles for biofluids (pharmacometabolomics) is also provided. Keywords: pharmacometabolomics; metabolomics; pharmacogenomics; therapeutic drug monitoring; personalized medicine; mass spectrometry 1. Introduction The uniformity of the drug response or low inter-individual differences in drug response are commonly accepted tenets in the field of medicine. Almost all drugs are prescribed on the basis of this statement. This approach can be described as treatment of the “average patient” by “the average pill” or “one size fits all”. However, clinicians have long observed that the actual effectiveness of the pharmacotherapy may be variable. -

The Metabolomic Paradigm of Pharmacogenomics in Complex
ics: O om pe ol n b A a c t c e e M s s Cacabelos, Metabolomics 2012, 2:5 Metabolomics: Open Access DOI: 10.4172/2153-0769.1000e119 ISSN: 2153-0769 Editorial Open Access The Metabolomic Paradigm of Pharmacogenomics in Complex Disorders Ramón Cacabelos1,2* 1EuroEspes Biomedical Research Center, Institute for CNS Disorders and Genomic Medicine, EuroEspes Chair of Biotechnology and Genomics, 15165-Bergondo, Corunna, Spain 2President, World Association of Genomic Medicine, Spain Metabolomics represents the networking organization of multiple pathogenic genes usually converge in metabolomic networks leading biochemical pathways leading to a physiological function in living to specific pathogenic cascades responsible for disease phenotypes. organisms. The frontier between health and disease is likely to be the The genomics of the mechanism of action of drugs has so far been result of a fine-tuning equilibrium or disequilibrium, respectively, neglected by the scientific community, and consequently less than 5% between the genomic-transcriptomic-proteomic-metabolomic cascade of FDA-approved drugs, with a pharmacologically-defined mechanism and environmental factors and/or epigenetic phenomena. In recent of action, have been studied in order to evaluate whether mutations times, diverse metabolomic studies have emerged in medical science in the genes encoding receptors or enzymes may affect efficacy and to explain physiological and pathogenic events in several disciplines, safety issues. Pleiotropic genes, involved in multiple pathogenic events, -
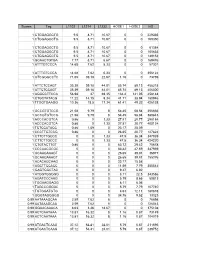
Scores Tag L1102 L1214 L1232 HOSE1 HOSE2 HS 1
Scores Tag L1102 L1214 L1232 HOSE1 HOSE2 HS 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 229335 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 169350 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 61384 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 105633 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 149152 1 GCAACTGTGA 7.77 8.71 6.67 0 0 169476 1 ATTTGTCCCA 14.68 7.62 5.33 0 0 57301 1 ATTTGTCCCA 14.68 7.62 5.33 0 0 356122 1 GTCGGGCCTC 71.65 39.18 22.67 1.16 0 73769 1 ATTCTCCAGT 35.39 39.18 44.01 85.74 89.13 458218 1 ATTCTCCAGT 35.39 39.18 44.01 85.74 89.13 406300 1 AGGGCTTCCA 56.98 37 69.35 134.4 141.05 458148 1 CTGCTATACG 11.22 14.15 9.34 41.71 38.94 180946 1 TTGGTGAAGG 10.36 18.5 17.34 61.41 49.32 426138 1 GCCGTGTCCG 21.58 9.79 8 54.45 58.84 356666 1 GCCGTGTCCG 21.58 9.79 8 54.45 58.84 380843 1 ACCCACGTCA 0.86 0 1.33 27.81 20.77 298184 1 ACCCACGTCA 0.86 0 1.33 27.81 20.77 400124 1 TCTCCATACC 0.86 1.09 0 23.17 25.09 1 CCCTTGTCCG 0.86 0 0 26.65 20.77 127824 1 CTTCTTGCCC 0 0 1.33 47.5 36.34 347939 1 CTTCTTGCCC 0 0 1.33 47.5 36.34 424220 1 CTGTACTTGT 0.86 0 0 63.72 29.42 75678 1 CCCAACGCGC 0 0 0 83.42 47.59 347939 1 GCAAGAAAGT 0 0 0 26.65 39.81 36977 1 GCAAGAAAGT 0 0 0 26.65 39.81 155376 1 ACACAGCAAG 0 0 0 23.17 15.58 1 AGCTTCCACC 0 0 0 11.59 7.79 355542 1 GAGTGGCTAC 0 0 0 9.27 6.92 1 ATGGTGGGGG 0 0 0 8.11 22.5 343586 1 AGATCCCAAG 0 0 0 5.79 8.65 50813 1 TGGAAGGAGG 0 0 0 8.11 6.06 1 TAGCCGGGAC 0 0 0 5.79 7.79 107740 1 TGTGGATGTG 0 0 0 4.63 12.11 180878 1 GGGTAGGGGG 0 0 0 34.76 9.52 13323 0.99 AATAAAGCAA 2.59 7.62 8 0 0 76698 0.99 AATAAAGCAA 2.59 7.62 8 0 0 126043 0.99 GGAACAAACA 8.63 3.26 18.67 0 0 375108 -

Model-Based Integration of Genomics and Metabolomics Reveals SNP Functionality in Mycobacterium Tuberculosis
Model-based integration of genomics and metabolomics reveals SNP functionality in Mycobacterium tuberculosis Ove Øyåsa,b,1, Sonia Borrellc,d,1, Andrej Traunerc,d,1, Michael Zimmermanne, Julia Feldmannc,d, Thomas Liphardta,b, Sebastien Gagneuxc,d, Jörg Stellinga,b, Uwe Sauere, and Mattia Zampierie,2 aDepartment of Biosystems Science and Engineering, ETH Zurich, 4058 Basel, Switzerland; bSIB Swiss Institute of Bioinformatics, 1015 Lausanne, Switzerland; cDepartment of Medical Parasitoloy and Infection Biology, Swiss Tropical and Public Health Institute, 4051 Basel, Switzerland; dUniversity of Basel, 4058 Basel, Switzerland; and eInstitute of Molecular Systems Biology, ETH Zurich, 8093 Zurich, Switzerland Edited by Ralph R. Isberg, Tufts University School of Medicine, Boston, MA, and approved March 2, 2020 (received for review September 12, 2019) Human tuberculosis is caused by members of the Mycobacterium infection of macrophages (29–32). Beyond analyses of individual tuberculosis complex (MTBC) that vary in virulence and transmis- laboratory strains, however, no systematic characterization and sibility. While genome-wide association studies have uncovered comparative analysis of intrinsic metabolic differences across several mutations conferring drug resistance, much less is known human-adapted MTBC clinical strains has been performed. about the factors underlying other bacterial phenotypes. Variation If the metabolic and other phenotypic diversity between in the outcome of tuberculosis infection and diseases has been MTBC strains contributes to and modulates pathogenicity, an attributed primarily to patient and environmental factors, but obvious question is: Which elements of the limited genetic di- recent evidence indicates an additional role for the genetic diver- versity in the MTBC are responsible for phenotypic strain di- sity among MTBC clinical strains. -

M. Tomita T. Nishioka (Eds.) Metabolomics the Frontier of Systems Biology
M. Tomita T. Nishioka (Eds.) Metabolomics The Frontier of Systems Biology M. Tomita, T. Nishioka (Eds.) Metabolomics The Frontier of Systems Biology With 112 Figures, Including 4 in Color Springer Masaru Tomita, Ph.D. Professor and Director Institute for Advanced Biosciences Keio University Tsuruoka 997-0035, Japan Takaaki Nishioka, Ph.D. Professor Graduate School of Agricuhure Kyoto University Kyoto 606-8502, Japan This book is based on the Japanese original, M. Tomita, T. Nishioka (Eds.), Metabolome Kenkyu no Saizensen, Springer-Verlag Tokyo, 2003. Library of Congress Control Number: 2005928331 ISBN 4-431-25121-9 Springer-Verlag Tokyo Berlin Heidelberg New York This work is subject to copyright. All rights are reserved, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in other ways, and storage in data banks. The use of registered names, trademarks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. Product liability: The publishers cannot guarantee the accuracy of any information about dosage and application contained in this book. In every individual case the user must check such information by consulting the relevant literature. Springer is a part of Springer Science+Business Media springeronline.com © Springer-Vertag Tokyo 2005 Printed in Japan Typesetting: Camera-ready by the editor. Printing and binding: Nikkei Printing, Japan Printed on acid-free paper Preface The aim of this book is to review metabolomics research. -

YEASTRACT - Genes Grouped by TF, Ordered by the Percentage of Genes Regulated by TF, Relative to the Total Number of Genes in the List 08/07/14 16:17
YEASTRACT - Genes grouped by TF, ordered by the percentage of genes regulated by TF, relative to the total number of genes in the list 08/07/14 16:17 Home > Group Genes by TF > Result Contact Us - Tutorial - Tutorial Genes grouped by TF, ordered by the percentage of genes regulated by TF, relative to the total number of genes in the list Quick search... Search Documented regulations suported by direct or indirect or undefined evidence. Unknown gene/ORF name(s), 'YFL013W-A'. DISCOVERER Transcription Transcriptional Regulatory % ORF/Genes Regulatory Associations: Factor Network - Search for TFs NTG1 YAL045c HAP3 - Search for Genes ATP1 YBL107c ALG1 - Search for Associations YSA1 CNS1 PDB1 TSC10 RER1 YCL049c PRD1 Group genes: AHC2 GET3 BPL1 - Group by TF YDL144c GCV1 PST2 - Group by GO PST1 SED1 YDR262w YDR266c MSW1 FRQ1 Pattern Matching: TSA2 STP1 PHO8 RAD23 - Search by DNA Motif YEL047c CAN1 YEL074w - Find TF Binding Site(s) PMI40 FMP52 CEM1 - Search Motifs on Motifs SER3 SHC1 YER130c Utilities: PDA1 YER189w AGP3 - ORF List ⇔ Gene List DUG1 CDH1 OCH1 - IUPAC Code Generation YGL114w MRM2 CHO2 - Generate Regulation Matrix PMT6 TRX2 YSC84 CHS7 Yap1p 24.9 % YSP1 FAA3 LYS1 GTT1 Retrieve: YJL045w GZF3 LCB3 - Transcription Factors List HXT9 VPS55 HOM6 BAT2 - Upstream Sequence THI11 RSM22 UBA1 - Flat files RHO4 PAM17 YKR070w YKR077w GTT2 YLL067c About Yeastract: ALT1 HOG1 MAS1 - Contact Us ECM38 ILV5 MRPL4 - Cite YEASTRACT TSA1 PRE8 TAF13 VAN1 - Acknowledgments YTA12 NUP53 SPG5 - Credits MRPL24 NIS1 APP1 CPT1 FMP41 MRPL19 YNL208w CWC25 LYP1 TOF1