Mini-Haplotypes As Lineage Informative Snps and Ancestry Inference Snps
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2012/174282 A2 20 December 2012 (20.12.2012) P O P C T (51) International Patent Classification: David [US/US]; 13539 N . 95th Way, Scottsdale, AZ C12Q 1/68 (2006.01) 85260 (US). (21) International Application Number: (74) Agent: AKHAVAN, Ramin; Caris Science, Inc., 6655 N . PCT/US20 12/0425 19 Macarthur Blvd., Irving, TX 75039 (US). (22) International Filing Date: (81) Designated States (unless otherwise indicated, for every 14 June 2012 (14.06.2012) kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, English (25) Filing Language: CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, Publication Language: English DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, (30) Priority Data: KZ, LA, LC, LK, LR, LS, LT, LU, LY, MA, MD, ME, 61/497,895 16 June 201 1 (16.06.201 1) US MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, 61/499,138 20 June 201 1 (20.06.201 1) US OM, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SC, SD, 61/501,680 27 June 201 1 (27.06.201 1) u s SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, 61/506,019 8 July 201 1(08.07.201 1) u s TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Content Based Search in Gene Expression Databases and a Meta-Analysis of Host Responses to Infection
Content Based Search in Gene Expression Databases and a Meta-analysis of Host Responses to Infection A Thesis Submitted to the Faculty of Drexel University by Francis X. Bell in partial fulfillment of the requirements for the degree of Doctor of Philosophy November 2015 c Copyright 2015 Francis X. Bell. All Rights Reserved. ii Acknowledgments I would like to acknowledge and thank my advisor, Dr. Ahmet Sacan. Without his advice, support, and patience I would not have been able to accomplish all that I have. I would also like to thank my committee members and the Biomed Faculty that have guided me. I would like to give a special thanks for the members of the bioinformatics lab, in particular the members of the Sacan lab: Rehman Qureshi, Daisy Heng Yang, April Chunyu Zhao, and Yiqian Zhou. Thank you for creating a pleasant and friendly environment in the lab. I give the members of my family my sincerest gratitude for all that they have done for me. I cannot begin to repay my parents for their sacrifices. I am eternally grateful for everything they have done. The support of my sisters and their encouragement gave me the strength to persevere to the end. iii Table of Contents LIST OF TABLES.......................................................................... vii LIST OF FIGURES ........................................................................ xiv ABSTRACT ................................................................................ xvii 1. A BRIEF INTRODUCTION TO GENE EXPRESSION............................. 1 1.1 Central Dogma of Molecular Biology........................................... 1 1.1.1 Basic Transfers .......................................................... 1 1.1.2 Uncommon Transfers ................................................... 3 1.2 Gene Expression ................................................................. 4 1.2.1 Estimating Gene Expression ............................................ 4 1.2.2 DNA Microarrays ...................................................... -
SUPPLEMENTARY DATA Supplementary Table 1. Characteristics of the Organ Donors and Human Islet Preparations Used for RNA-Seq
SUPPLEMENTARY DATA Supplementary Table 1. Characteristics of the organ donors and human islet preparations used for RNA-seq and independent confirmation and mechanistic studies. Gender Age BMI Cause of death Purity (years) (kg/m2) (%) F 77 23.8 Trauma 45 M 36 26.3 CVD 51 M 77 25.2 CVD 62 F 46 22.5 CVD 60 M 40 26.2 Trauma 34 M 59 26.7 NA 58 M 51 26.2 Trauma 54 F 79 29.7 CH 21 M 68 27.5 CH 42 F 76 25.4 CH 30 F 75 29.4 CVD 24 F 73 30.0 CVD 16 M 63 NA NA 46 F 64 23.4 CH 76 M 69 25.1 CH 68 F 23 19.7 Trauma 70 M 47 27.7 CVD 48 F 65 24.6 CH 58 F 87 21.5 Trauma 61 F 72 23.9 CH 62 M 69 25 CVD 85 M 85 25.5 CH 39 M 59 27.7 Trauma 56 F 76 19.5 CH 35 F 50 20.2 CH 70 F 42 23 CVD 48 M 52 24.5 CH 60 F 79 27.5 CH 89 M 56 24.7 Cerebral ischemia 47 M 69 24.2 CVD 57 F 79 28.1 Trauma 61 M 79 23.7 NA 13 M 82 23 CH 61 M 32 NA NA 75 F 23 22.5 Cardiac arrest 46 M 51 NA Trauma 37 Abbreviations: F: Female; M: Male; BMI: Body mass index; CVD: Cardiovascular disease; CH: Cerebral hemorrhage. -

Med1 Regulates Meiotic Progression During Spermatogenesis in Mice
REPRODUCTIONRESEARCH Differences in the transcriptional profiles of human cumulus cells isolated from MI and MII oocytes of patients with polycystic ovary syndrome Xin Huang, Cuifang Hao, Xiaofang Shen, Xiaoyan Liu, Yinghua Shan, Yuhua Zhang and Lili Chen Reproductive Medicine Centre, Yuhuangding Hospital of Yantai, Affiliated Hospital of Qingdao Medical University, 20 Yuhuangding Road East, Yantai, Shandong, 264000, People’s Republic of China Correspondence should be addressed to C Hao; Email: [email protected] Abstract Polycystic ovary syndrome (PCOS) is a common endocrine and metabolic disorder in women. The abnormalities of endocrine and intra-ovarian paracrine interactions may change the microenvironment for oocyte development during the folliculogenesis process and reduce the developmental competence of oocytes in PCOS patients who are suffering from anovulatory infertility and pregnancy loss. In this microenvironment, the cross talk between an oocyte and the surrounding cumulus cells (CCs) is critical for achieving oocyte competence. The aim of our study was to investigate the gene expression profiles of CCs obtained from PCOS patients undergoing IVF cycles in terms of oocyte maturation by using human Genome U133 Plus 2.0 microarrays. A total of 59 genes were differentially expressed in two CC groups. Most of these genes were identified to be involved in one or more of the following pathways: receptor interactions, calcium signaling, metabolism and biosynthesis, focal adhesion, melanogenesis, leukocyte transendothelial migration, Wnt signaling, and type 2 diabetes mellitus. According to the different expression levels in the microarrays and their putative functions, six differentially expressed genes (LHCGR, ANGPTL1, TNIK, GRIN2A, SFRP4, and SOCS3) were selected and analyzed by quantitative RT-PCR (qRT-PCR). -

Hepatocyte Differentiation and Hepatocellular Carcinoma
HEPATOCYTE DIFFERENTIATION AND HEPATOCELLULAR CARCINOMA: RATIONALE FOR P53 INDEPENDENT THERAPY by FRANCIS O ENANE Submitted in partial fulfillment of the requirement for the degree of Doctor of Philosophy Dissertation Advisor Yogen Saunthararajah, MD Department of Molecular Medicine Cleveland Clinic Lerner College of Medicine CASE WESTERN RESERVE UNIVERSITY May 2017 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the dissertation of Francis O Enane Candindate for Doctor of Philosophy Degree*. Committee Chair: Peter Scacheri, PhD Committee member: Angela Ting PhD Committee member: Xiaoxia Li, PhD Committee member: Alok Khorana, MD Committee member: Yogen Saunthararajah, MD Date of defense: December 19th 2016 *We also certify that written approval has been obtained for any proprietary material contained therein Dedication I dedicate this work to approximately 17.5 million global cancer patient population as of the year 2016. I strongly believe that the scientific and medical communities will continue to work coherently to identify mechanisms to provide better cure rates of cancer, to reduce the economic burden to families affected, and to define psychological and emotional challenges experienced by patients and their families. The work performed in this dissertation is a small contribution to that objective and paves the way to understand new therapeutic mechanisms in hepatocellular carcinoma. In the modern technical and highly skilled society - and with sufficient financial and political support - there will -

A Genome-Wide Association Study of Serum Proteins Reveals Shared Loci with Common Diseases
bioRxiv preprint doi: https://doi.org/10.1101/2021.07.02.450858; this version posted July 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. 1 A genome-wide association study of serum proteins reveals shared loci with common 2 diseases 3 Alexander Gudjonsson*,1, Valborg Gudmundsdottir*,1,2, Gisli T Axelsson1,2, Elias F 4 Gudmundsson1, Brynjolfur G Jonsson1, Lenore J Launer3, John R Lamb4, Lori L Jennings5, Thor 5 Aspelund1,2, Valur Emilsson#,1,2 & Vilmundur Gudnason#,1,2 6 7 8 9 1Icelandic Heart Association, Holtasmari 1, 201 Kopavogur, Iceland. 10 2Faculty of Medicine, University of Iceland, 101 Reykjavik, Iceland. 11 3Laboratory of Epidemiology and Population Sciences, Intramural Research Program, National 12 Institute on Aging, Bethesda, MD 20892-9205, USA. 13 4GNF Novartis, 10675 John Jay Hopkins Drive, San Diego, CA 92121, USA. 14 5Novartis Institutes for Biomedical Research, 22 Windsor Street, Cambridge, MA 02139, USA. 15 16 17 18 19 *These authors contributed equally as joint-first authors 20 #These authors contributed equally as joint-senior authors 21 22 23 24 25 26 27 28 Correspondence: [email protected] 29 Keywords: Proteomics, pQTLs, genomics, systems genetics, serum 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.07.02.450858; this version posted July 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Application of Minimally-Invasive Uterine Fluid Aspiration to Identify Candidate Biomarkers of Endometrial Receptivity Through a Transcriptomic Approach
Application of Minimally-Invasive Uterine Fluid Aspiration to Identify Candidate Biomarkers of Endometrial Receptivity through a Transcriptomic Approach by Crystal Chan A thesis submitted in conformity with the requirements for the degree of Master of Science Institute of Medical Science University of Toronto © Copyright by Crystal Chan 2012 Application of Minimally-Invasive Uterine Fluid Aspiration to Identify Candidate Biomarkers of Endometrial Receptivity through a Transcriptomic Approach Crystal Chan Master of Science Institute of Medical Science University of Toronto 2012 Abstract The endometrium is receptive to the embryo during a restricted window in the mid-secretory phase. My objectives were to develop a minimally-invasive endometrial sampling method for gene expression profiling, and to identify genes differentially expressed in the receptive phase. Twenty-three normo-ovulatory women underwent uterine fluid aspiration during the pre- receptive (LH+2) and receptive (LH+7) phase of the same natural cycle. RNA was extracted, reverse transcribed, amplified and hybridized to whole-genome microarrays. Unsupervised hierarchical clustering revealed self-segregation of pre-receptive and receptive samples. Importantly, profiling by uterine fluid aspiration was representative of biopsy. An unpaired t-test with a false discovery rate of 0.05 and a Δ threshold of 4-fold identified 245 unique transcripts as differentially expressed in the receptive phase. NanoString analysis validated 96% of these genes. This approach will now allow us to correlate expression of these candidate biomarkers to implantation outcomes, towards the development of clinical assays predictive for endometrial receptivity. ii Acknowledgments I would first and foremost like to thank Dr. Ted Brown and Dr. Ellen Greenblatt, supervisors extraordinaire, who gave me the resources, inspiration, and unconditional support to transform this research from a pipe dream to reality. -

Revelation of Pivotal Genes Pertinent to Alzheimer's Pathogenesis
Revelation of pivotal genes pertinent to Alzheimer’s pathogenesis: A methodical evaluation of 32 GEO datasets Hema Sree G N S M. S. Ramaiah University of Applied Sciences Saraswathy Ganesan Rajalekshmi ( [email protected] ) M. S. Ramaiah University of Applied Sciences Raghunadha R Burri Dr. Reddy’s Laboratories Research Article Keywords: BDNF, SST, SERPINA3, RTN3, RGS4 Posted Date: May 20th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-533893/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/40 Abstract Background: Alzheimer’s Disease (AD), a dreadful neurodegenerative disorder that affects cognitive and behavioral functions in geriatric population, is characterized by the presence of amyloid deposits and neurobrillary tangles in hippocampus and cortex regions of the BRAIN. The 2018 World Alzheimer’s Report exemplied a global prevalence of 50 million AD cases and forecasted a threefold rise upto 2 trillion by 2050. Although, there exist numerous genetic association studies pertinent to ad in different ethnicities, yet, critical genetic factors and signaling pathways underlying its pathogenesis still remain ambiguous. This study was aimed to collate and analyze the genetic data retrieved from 32 Gene Expression Omnibus (GEO) datasets belonging to diverse ethnic cohorts in order to identify overlapping Differentially Expressed Genes (DEGs). Stringent selection criteria were framed to shortlist appropriate datasets based on FDR p-value, log FC and relevant details on expression of both upregulated and downregulated DEGs from the same dataset. Results: Among the 32 datasets, only 6 datasets satised the selection criteria. Geo2R tool was SUBSEQUENTLY employed to retrieve signicant DEGs. -

Application of Custom-Designed Oligonucleotide Array CGH in 145 Patients with Autistic Spectrum Disorders
European Journal of Human Genetics (2013) 21, 620–625 & 2013 Macmillan Publishers Limited All rights reserved 1018-4813/13 www.nature.com/ejhg ARTICLE Application of custom-designed oligonucleotide array CGH in 145 patients with autistic spectrum disorders Barbara Wis´niowiecka-Kowalnik1, Monika Kastory-Bronowska2, Magdalena Bartnik1, Katarzyna Derwin´ska1, Wanda Dymczak-Domini2, Dorota Szumbarska2, Ewa Ziemka2, Krzysztof Szcza$uba1, Maciej Sykulski3, Tomasz Gambin4, Anna Gambin3,5, Chad A Shaw6, Tadeusz Mazurczak1,7, Ewa Obersztyn1, Ewa Bocian1 and Pawe$ Stankiewicz*,1,6 Autism spectrum disorders (ASDs) are a heterogeneous group of neurodevelopmental disorders, including childhood autism, atypical autism, and Asperger syndrome, with an estimated prevalence of 1.0–2.5% in the general population. ASDs have a complex multifactorial etiology, with genetic causes being recognized in only 10–20% of cases. Recently, copy-number variants (CNVs) have been shown to contribute to over 10% of ASD cases. We have applied a custom-designed oligonucleotide array comparative genomic hybridization with an exonic coverage of over 1700 genes, including 221 genes known to cause autism and autism candidate genes, in a cohort of 145 patients with ASDs. The patients were classified according to ICD-10 standards and the Childhood Autism Rating Scale protocol into three groups consisting of 45 individuals with and 69 individuals without developmental delay/intellectual disability (DD/ID), and 31 patients, in whom DD/ID could not be excluded. In 12 patients, we have identified 16 copy-number changes, eight (5.5%) of which likely contribute to ASDs. In addition to known recurrent CNVs such as deletions 15q11.2 (BP1-BP2) and 3q13.31 (including DRD3 and ZBTB20), and duplications 15q13.3 and 16p13.11, our analysis revealed two novel genes clinically relevant for ASDs: ARHGAP24 (4q21.23q21.3) and SLC16A7 (12q14.1). -

IJHOSCR Original Article International Journal of Hematology-Oncology and Stem Cell Research
Archive of SID IJHOSCR Original Article International Journal of Hematology-Oncology and Stem Cell Research Transcriptomic Profiles of MV4-11 and Kasumi 1 Acute Myeloid Leukemia Cell Lines Modulated by Epigenetic Modifiers Trichostatin A and 5- Azacytidine Mat Jusoh Siti Asmaa, Hamid Ali Al-Jamal, Abdul Rahim Hussein, Badrul Hisham Yahaya, Azlan Husin, Roslin Hassan1, Faezahtul Arbaeyah Hussain5, Shaharum Shamsuddin6,7, Muhammad Farid Johan1 1Department of Hematology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia 2Diagnostic and Biomedicine, Faculty of Health Sciences, Universiti Sultan Zainal Abidin, Gong Badak Campus, Kuala Nerus, 21300, Terengganu, Malaysia 3Regenerative Medicine Cluster, Advanced Medical and Dental Institute, Universiti Sains Malaysia, Bertam, 13200 Kepala Batas, Pulau Pinang, Malaysia 4Department of Internal Medicine, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia 5Department of Pathology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia 6School of Health Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia 7Institute for Research in Molecular Medicine (INFORMM), Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia Corresponding Author: Muhammad Farid Johan, Department of Hematology, School of Medical Sciences, Universiti Sains Malaysia, 16150 Kubang Kerian, Kelantan, Malaysia. Tel: +60-13-9824550 Fax: +6097673333 Email: [email protected] Received: 14, Feb, 2019 Accepted: 27, Apr, 2019 ABSTRACT Background: Acute myeloid leukemia (AML) is the most common form of acute leukemias in adults which is clinically and molecularly heterogeneous. Several risk and genetic factors have been widely investigated to characterize AML. However, the concomitant epigenetic factors in controlling the gene expression lead to AML transformation was not fully understood. -
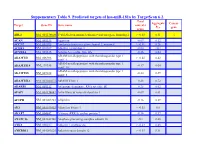
Suppementary Table 9. Predicted Targets of Hsa-Mir-181A by Targetscan 6.2
Suppementary Table 9. Predicted targets of hsa-miR-181a by TargetScan 6.2. Total Aggregate Cancer Target Gene ID Gene name context+ P gene score CT ABL2 NM_001136000 V-abl Abelson murine leukemia viral oncogene homolog 2 > -0.03 0.31 √ ACAN NM_001135 Aggrecan -0.09 0.12 ACCN2 NM_001095 Amiloride-sensitive cation channel 2, neuronal > -0.01 0.26 ACER3 NM_018367 Alkaline ceramidase 3 -0.04 <0.1 ACVR2A NM_001616 Activin A receptor, type IIA -0.16 0.64 ADAM metallopeptidase with thrombospondin type 1 ADAMTS1 NM_006988 > -0.02 0.42 motif, 1 ADAM metallopeptidase with thrombospondin type 1 ADAMTS18 NM_199355 -0.17 0.64 motif, 18 ADAM metallopeptidase with thrombospondin type 1 ADAMTS5 NM_007038 -0.24 0.59 motif, 5 ADAMTSL1 NM_001040272 ADAMTS-like 1 -0.26 0.72 ADARB1 NM_001112 Adenosine deaminase, RNA-specific, B1 -0.28 0.62 AFAP1 NM_001134647 Actin filament associated protein 1 -0.09 0.61 AFTPH NM_001002243 Aftiphilin -0.16 0.49 AK3 NM_001199852 Adenylate kinase 3 > -0.02 0.6 AKAP7 NM_004842 A kinase (PRKA) anchor protein 7 -0.16 0.37 ANAPC16 NM_001242546 Anaphase promoting complex subunit 16 -0.1 0.66 ANK1 NM_000037 Ankyrin 1, erythrocytic > -0.03 0.46 ANKRD12 NM_001083625 Ankyrin repeat domain 12 > -0.03 0.31 ANKRD33B NM_001164440 Ankyrin repeat domain 33B -0.17 0.35 ANKRD43 NM_175873 Ankyrin repeat domain 43 -0.16 0.65 ANKRD44 NM_001195144 Ankyrin repeat domain 44 -0.17 0.49 ANKRD52 NM_173595 Ankyrin repeat domain 52 > -0.05 0.7 AP1S3 NM_001039569 Adaptor-related protein complex 1, sigma 3 subunit -0.26 0.76 Amyloid beta (A4) precursor protein-binding, family A, APBA1 NM_001163 -0.13 0.81 member 1 APLP2 NM_001142276 Amyloid beta (A4) precursor-like protein 2 -0.05 0.55 APOO NM_024122 Apolipoprotein O -0.32 0.41 ARID2 NM_152641 AT rich interactive domain 2 (ARID, RFX-like) -0.07 0.55 √ ARL3 NM_004311 ADP-ribosylation factor-like 3 > -0.03 0.51 ARRDC3 NM_020801 Arrestin domain containing 3 > -0.02 0.47 ATF7 NM_001130059 Activating transcription factor 7 > -0.01 0.26 ATG2B NM_018036 ATG2 autophagy related 2 homolog B (S. -

Symbol- Und Abkürzungsverzeichnis
Untersuchung des Beitrags von ETV6/RUNX1 zur Entstehung akuter lymphatischer Leukämie (ALL) im Kindesalter Inauguraldissertation zur Erlangung des Grades eines Doktors der Naturwissenschaften (Dr. rer. nat.) im Fachbereich Biologie, Chemie und Pharmazie der Freien Universität Berlin vorgelegt von Kerstin Hasse Berlin, Dezember 2012 1. Gutachter: Prof. Hartmut Oschkinat Forschungsinstitut für Molekulare Pharmakologie, Berlin 2. Gutachter: Prof. Burghardt Wittig Institut für Molekularbiologie und Bioinformatik der FU Berlin Tag der Disputation: 19. November 2012 Inhaltsverzeichnis Symbol- und Abkürzungsverzeichnis ...................................................I 1. Einleitung und Zielsetzung ................................................................1 1.1. Leukämien ................................................................................................... 1 1.2. ALL im Kindesalter....................................................................................... 2 1.2.1. Immunphänotypen der ALL........................................................... 3 1.2.2. Zytogenetische und molekulargenetische Veränderungen bei ALL ...................................................................................................... 4 1.3. Molekulare Pathogenese von Leukämien.................................................... 7 1.4. Die Transkriptionsfaktoren ETV6 und RUNX1 ............................................. 8 1.4.1. Transkriptionsfaktor ETV6............................................................. 8 1.4.1.1.