Methods in Molecular Biology 1037
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Basic Function of the Anesthetic Machine, Part II Some Vaporizers Are Filled with Liquid Agent by Using a Pin Fill Device
Vetamac Vapors (800)334-1583 www.vetamac.com Vol I, Issue 2 Welcome to the second quarterly edition of "Vetamac Vapors". The purpose of this newsletter is to educate your staff with a goal of the best possible patient care. We also would like to inform you of services and products that Vetamac has to offer and to provide support for all of your anesthesia needs. Future editions will cover topics on machines as well as different techniques. Please send comments to the e-mail link at www.vetamac.com. Basic Function of the Anesthetic Machine, Part II Some vaporizers are filled with liquid agent by using a pin fill device. There is an agent specific spout that replaces the cap on the The anesthetic machine must deliver vaporized anesthetic agent bottle. There is a keyed pin that fits into the fill manifold on the agent to the breathing circuit in concentrations that are optimal for the vaporizer. It is locked into place and the vaporizer is filled to the de- sired level on the window. Most vaporizers used in the U.S. have a desired effect. Since the liquid agent is in a closed system, a carrier funnel fill device and the liquid agent is simply poured into the vapor- gas must be present to deliver the vaporized agent to the breathing izer. Care must be exercised to avoid spillage. Anti-Spil™ adapters circuit. The gas used to accomplish this is the fresh gas flow of oxy- are available that replace the cap on the agent bottle and minimize the gen. -

Total Solutions for Anesthesia & Surgery
total solutions for Anesthesia & Surgery Ventilation Anesthesia Animal Surgery Temperature Control Physiological Monitoring NEW Catalogs Other Catalogs from Harvard Apparatus 2010 BTX Electroporation Catalog BTX offers a comprehensive line of instruments and accessories for both electroporation and electrofusion of mammalian, bacterial, yeast, fungi, insect and plant cells and tissues. BTX specializes in providing research tools for novel cutting edge applications such as adherent cell electroporation, high- throughput cloning, in vivo gene delivery, in ovo gene delivery, and in & ex utero gene delivery. 2009 Warner 2010 Animal, Organ & Cell Physiology Electrophysiology and This catalog features a broad range of products, including our Cell Biology Catalog legendary line of infusion and perfusion pumps, ventilators and anesthesia systems, surgical instruments and equipment for small For the electrophysiological, cellular, and to larger animals, and isolated organ and tissue systems for all neurological sciences. Our extensive levels of research and education. These products are designed to product line includes voltage and current help you achieve better research results in less time. clamp amplifiers for whole-cell and patch applications. Bessel filters, chambers for imaging and recording systems, perfusion control systems and steppers, 2010 Molecular solution heating systems, microscope translation tables, microelectrodes and holders, and glass capillary tubing, plus Sample Preparation now includes electroporation and transfection systems -
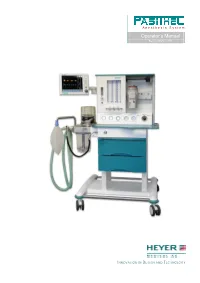
HEYER Pasithec
Anesthesia System Operator’s Manual Rev. 0.2 Draft – 12/09 INNOVATION IN DESIGN AND TECHNOLOGY HEYER Pasithec Contents 1 Statement .......................................................................................................................................................................... 5 1.1 Manufacturer Responsibility...................................................................................................................................... 5 1.2 Security, Reliability and Operating Conditions........................................................................................................... 5 1.3 Return ...................................................................................................................................................................... 6 1.4 Details of the Manufacturer....................................................................................................................................... 6 2 Introduction........................................................................................................................................................................ 7 2.1 Intended Use............................................................................................................................................................ 7 2.1.1 Range of Use .................................................................................................................................................. 7 2.1.2 Contraindication ............................................................................................................................................. -

CORRESPONDENCE Does Restrictive
Ⅵ CORRESPONDENCE Anesthesiology 2006; 104:889 © 2006 American Society of Anesthesiologists, Inc. Lippincott Williams & Wilkins, Inc. Does Restrictive Intraoperative Fluid Management Alter Outcome after Intraabdominal Surgery? To the Editor:—I read with great interest the recent article by Nisanev- confidence interval includes 1 and so would not be taken to indicate a ich et al.1 suggesting that restricted fluid therapy for intraabdominal statistical difference between the two therapies. surgery reduces postoperative morbidity. My comments focus on the The authors mention the use of the Newman-Keuls adjustment, but description of the statistical methods and their application to the data. that correction only applies if the group means are independent, The authors mention the use of both the chi-square test and Fisher which is clearly not the case here. Downloaded from http://pubs.asahq.org/anesthesiology/article-pdf/104/4/890/361043/0000542-200604000-00040.pdf by guest on 02 October 2021 exact test for the analysis of categorical data. However, it seems that No advanced statistical methods are used to model the data and only the results for the chi-square test are reported: For the number of explain the impact of relevant covariates. In particular, logistic regres- patients with complications, the Fisher exact test gives a nonsignificant sion could be used to model the presence of a complication on the P value of 0.056. The authors should explain why they report the number of fluid boluses, the degree of hypotension, the duration of results of one test and not the other. surgery, or American Society of Anesthesiologists physical status. -

Interruption in the Supply of Breathing Gas During General Anesthesia Due to Malposition of the Vaporizer -A Case Report
Korean J Anesthesiol 2010 October 59(4): 270-274 Case Report DOI: 10.4097/kjae.2010.59.4.270 Interruption in the supply of breathing gas during general anesthesia due to malposition of the vaporizer -A case report- Hyo Jin Kim, and Mi Woon Kim Department of Anesthesiology and Pain Medicine, College of Medicine, Dongguk University, Gyeongju, Korea We report a case of interruption in the supply of breathing gas during general anesthesia caused by malposition of the Drager Vapor 2000Ⓡ vaporizer, which was accidentally tilted and lifted off the Selectatec manifold of the anesthesia machine. Because the patient was an 1-month-old infant, we couldn't check if he had experienced awareness with recall. We emphasize the importance of checking the anesthetic vaporizer after mounting it on the back bar of the anesthesia machine. (Korean J Anesthesiol 2010; 59: 270-274) Key Words: Gas leak, Selectatec, Vaporizer. The Selectatec, a vaporizer mounting system designed We experienced a case in which the Drager Vapor 2000Ⓡ by Datex-Ohmeda has several advantages over permanent vaporizer was incorrectly mounted on the Selectatec manifold mounting systems. It allows the vaporizer to be easily mounted, of the Datex-Ohmeda AestivaⓇ/5 compact plus anesthesia removed, and replaced, even during a case. The anesthesia machine, which caused a gas leak and interruption in the machine can have fewer mounting locations, allowing a more supply of breathing gas. compact machine. If malignant hyperthermia is suspected, the vaporizers can be removed. This gives better results than Case Report if the vaporizers remain on the machine in the off position [1]. -

Methods for Minimizing Pain Flare-Ups Survey
Methods for Minimizing Pain Flare‐Ups Survey I tried aqua therapy in a warm pool this past year. It helps to bypass my movement disorder. Not much minimizes my pain. I figure I'm going to be in pain doing nothing, and if I'm doing something. So I keep on keeping on. I decided to get a Harley Davidson a few years back and I ride to desensitize my nerves in my legs and helps to take my mind off it. Stay active regardless of the pain and reflect on what you accomplished instead of what you didn't. Even the smallest of things deserve your praise. Ketamine infusions are my monthly routine and I have them done now in the comfort of my own home. Water therapy is my biggest source of non‐medicinal pain relief. Moving around Warm weather, water therapy, and keeping busy to keep my mind off the pain. Also magnesium supplements to help me sleep better. Lavender lotion at night. Plenty of rest not just sleep but relax and rest throughout the day. It’s not just one thing that helps but a lot of little things. Don’t let the pain stop you although it may slow you down. Pain meds, antispasmodic pills, depression pills, anxiety pills, taking it easy not overdoing it, accepting it. Trusting God through each and every day. Nothing SO far, but I keep on keeping on the BEST I can I have found that two methods work best for me. I use a light therapy device that I actually bought from an infomercial when I was in desperate pain. -

SOP 1102 Surgivet® Anesthesia Machine
STANDARD OPERATING PROCEDURES DIVISION OF COMPARATIVE MEDICINE UNIVERSITY OF SOUTH FLORIDA SOP#: 1102.5 Date Issued: 5/99 Date Revised: 5/16 Page 1 of 3 TITLE: SurgiVet ™ Anesthesia Machine SCOPE: Research and Animal Care Personnel, RESPONSIBILITY: Facility Manager, Technical Staff, and Professional & Administrative Staff PURPOSE: To Outline the Proper Procedures for Use and Maintenance of Veterinary Inhalation Anesthesia Equipment I. PURPOSE 1. Isoflurane inhalation provides safe general anesthesia for a variety of animal species. This procedure outlines the use and maintenance of a veterinary inhalation anesthesia machine that incorporates an oxygen flowmeter, anesthetic vaporizer, and a circle re-breathing system with carbon dioxide absorption. II. RESPONSIBILITY 1. The Facility Manager ensures that equipment is appropriately cleaned, maintained in good working order, and available for research personnel as requested. 2. The veterinary professional, administrative, and managerial staff ensure that all research and technical staff using this equipment are adequately trained and experienced to perform veterinary inhalant general anesthesia. 3. The veterinary and technical staff operating this equipment ensure this procedure and the manufacturer’s operation manual are followed. III. EQUIPMENT SET- UP 1. Check that machine is connected to oxygen supply that is turned on and has an adequate supply at 50-55 psi. 2. Check the operation of the flow meter. 3. Check that vaporizer is adequately filled. 4. Check breathing circuit configuration and connections. 5. Check the scavenger system. Charcoal canisters should be weighed and replaced as recommended by the manufacturer. 6. Check ventilator function and connections. 7. Pressure test the machine as described in the operation manual. -

American Board of Anesthesiology American
AMERICAN BOARD OF ANESTHESIOLOGY AMERICAN SOCIETY OF ANESTHESIOLOGISTS IN-TRAINING EXAMINATION Book A 1993 PREPARED IN COOPERATION WITH NATIONAL BOARD OF MEDICAL EXAMINERS® Copyright© 1993 by the Joint Council on In-Training Examinations, American Board of Anesthesiology/American Society of Anesthesiologists. All rights Reserved. Reproduction of this Copyrighted Material is Prohibited. DIRECTIONS : Each of the numbered items or incomplete statements in this section is followed by answers or by comple• tions of the statement. Select the ONE lettered answer or completion that is BEST in each case and fill in the circle contain• ing the corresponding letter on the answer sheet. 1. Left ventricular subendocardial perfusion pressure is best estimated by the difference between (A) mean arterial and central venous pressures (B) diastolic arterial and pulmonary artery occlusion pressures (C) mean arterial and pulmonary artery occlusion pressures (D) systolic arterial and pulmonary artery occlusion pressures (E) diastolic arterial and central venous pressures 2. Oxygen 100 ml/min is bubbled through a vaporizer containing an anesthetic with a vapor pressure of 150 mmHg, and this mixture is added to a fresh gas flow of 5 L!min. The delivered anesthetic concentration is (A) 0.25% (B) 0.5% (C) 1% (D) 2.5% (E) 5% 3. In children with preoperative upper respiratory tract infection, which of the following is associated with the greatest risk for postoperative airway obstruction? (A) Age less than 1 year (B) Endotracheal intubation (C) Head and neck surgery (D) Inadequate airway humidification (E) Surgery for more than two hours 4. An 8-kg, 1-year-old child has a measured blood loss of 50 ml during the first two hours of a rectal pull-through opera• tion. -

Inhalation Anesthetics & Vaporizers
SCIENCE AND TECHNOLOGY Inhalation Anesthetics & Vaporizers The development and functionality of the anesthetic vaporizer and how Sevoflurane relates to its specific vaporizer model, physiological indication, and molecular structure. MICHAEL CRAIG, BS, AAS, CER.A.T.T. CONTRIBUTING EDITOR: BRYAN FULTON, BAA, CER.A.T.T. OKLAHOMA CITY COMMUNITY COLLEGE Historical Significance he origins of the anesthesia vaporizer are quite muddled; However, that is beyond this paper's purpose. It would appear Tfrom historical accounts that the first public demonstration of a physician utilizing a vaporizing apparatus to anesthetize a patient for the purposes of a surgical procedure occurred on October 16, 1846 (WLM, 2019). This monumental moment in the history of anesthesia took place at the Ether Dome, a surgical operating amphitheater located in the Bulfinch Building at Massachusetts General Hospital in Boston. The patient, Edward Gilbert Abbott, was undergoing a MORE The Sensor Winter 2020 SCIENCE AND TECHNOLOGY CONTINUING FROM PREVIOUS PAGE tumor mass resection procedure (WLM, 2019). The surgical team accomplished this feat with the use of Ether and a Morton inhaler [fig.1]. The Morton inhaler, designed by William T.G. Morton that same year, is considered the first vaporizer but was simply a container to hold the volatile anesthetic. Despite this momentous period in history, the science of anesthesia would have to advance beyond a glass container and the highly flammable and explosive Ether (Pharmacopoeia, 2016). Since 1846, there have been many scientific advances in volatile anesthetics. A primary advancement in general anesthetics revolves around the introduction of halogenated anesthetics. Halogenated anesthetics (HA) are currently the most widely used inhalation anesthetics in surgery today Figure 1: Morton Inhaler (NIH, 2019). -

Anesthetic Machine (Isoflurane) Operation
IACUC Standard Operating Procedure Anesthetic Machine (Isoflurane) Operation Objective: To describe use of the isoflurane vaporizer-style anesthetic machine Author: Chandra D. Williams, DVM, DACLAM, CPIA Date Written: Reviewed February 6, 2016 Approved: December 7, 2009; February 6, 2016 Introduction This Standard Operating Procedure will detail procedures to be followed when using the anesthesia machine for rodent survival and non-survival surgeries. It is recommended that the procedure room be reserved, in advance, by contacting the Vivarium at 704-687-8318. Procedure 1) Complete the Surgical Checklist and top portion of Animal Anesthesia Log. Confirm identity and protocol number of subject animal. 1) Turn the oxygen on by turning the valve in a counter-clockwise direction. Check the pressure gauge to make sure there is sufficient oxygen in the tank. Replace the tank immediately when pressure gauge registers less than 200 PSI. Vivarium staff may be contacted for replacement tanks. 2) Visually check the level of Isoflurane in the vaporizer. It should be at the half way mark to ensure that you do not run out of Isoflurane in the middle of a surgery. a) If the Isoflurane level is below the half way mark then additional Isoflurane is stored in the bottom of the cabinet in the treatment room. Unscrew the cap of the anesthesia reservoir. Remove stopper from Isoflurane and pour into vaporizer reservoir until the level reaches half way mark. Replace the reservoir cap. Place the stopper back on the Isoflurane and return it to the cabinet. Inform the Vivarium manager, or another member of the Vivarium staff, if Isoflurane is running low or is completely out. -

(12) United States Patent (10) Patent No.: US 9,655,867 B2 Spakevicius Et Al
US0096.55867B2 (12) United States Patent (10) Patent No.: US 9,655,867 B2 Spakevicius et al. (45) Date of Patent: May 23, 2017 (54) ANALGESC COMPOSITIONS (58) Field of Classification Search CPC .......... A61K 31/08: A61K 31/02: A61 K9/70; (71) Applicant: VAPOGENIX, INC., Houston, TX A61K 47/10; A61K 47/12: A61K 47/20; (US) A61K 47/34: A61K 47/38 (72) Inventors: Danguole Spakevicius, Houston, TX (Continued) (US); Heather Giles, Houston, TX (US); Terry Farmer, Houston, TX (56) References Cited (US) U.S. PATENT DOCUMENTS (73) Assignee: Vapogenix, Inc., Houston, TX (US) 7,736,537 B1 6/2010 Zastrow et al. (*) Notice: Subject to any disclaimer, the term of this 2011/O159078 A1 6, 2011 Burton et al. patent is extended or adjusted under 35 (Continued) U.S.C. 154(b) by 0 days. FOREIGN PATENT DOCUMENTS (21) Appl. No.: 14/775,584 GB 154.7225 A 6, 1979 WO 2008O36858 A2 3, 2008 (22) PCT Fed: Mar. 14, 2014 (Continued) (86) PCT No.: PCT/US2O14/028169 S 371 (c)(1), OTHER PUBLICATIONS (2) Date: Sep. 11, 2015 International Search Report (ISA/US) dated Sep. 30, 2014 for PCT PCT Pub. No.: WO2O14f143964 International Application No. PCT/US2014/028169, filed Mar. 14, (87) 2014. PCT Pub. Date: Sep. 18, 2014 (Continued) (65) Prior Publication Data Primary Examiner — T. Victor Oh US 2016/0022603 A1 Jan. 28, 2016 (74) Attorney, Agent, or Firm — Saul Ewing LLP: Related U.S. Application Data Kathryn Doyle; Domingos J. Silva (60) Provisional application No. 61/814,485, filed on Apr. (57) ABSTRACT 22, 2013, provisional application No. -

Operating Instructions
Operating Instructions ™ AnesthesiaANESTHESIA SYSTEM System Operating Instructions ™ ANESTHESIAAnesthesia SYSTEM System A7™ is an U.S. trademarks of Mindray DS USA, Inc. OPTIMIZER® is a registered trademark of Mindray DS USA, Inc. Selectatec® is a registered trademark of Ohmeda. Copyright . Mindray DS USA, Inc., 2013 to 2016. All rights reserved. Contents of this publication may not be reproduced in any form without permission of Mindray DS USA, Inc. Table of Contents Table of Contents Foreword ..............................................................................................................................................................................................................................ix Indications For Use ...........................................................................................................................................................................................................ix Responsibilities of Operators ........................................................................................................................................................................................ix Warnings, Cautions, and Notes ....................................................................................................................................................................................ix Warnings................................................................................................................................................................................................................................x