L-Helical Catalytic Domains in Glycoside Hydrolase Families 49, 55 and 87: Domain Architecture, Modelling and Assignment of Catalytic Residues
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Information
doi: 10.1038/nature06269 SUPPLEMENTARY INFORMATION METAGENOMIC AND FUNCTIONAL ANALYSIS OF HINDGUT MICROBIOTA OF A WOOD FEEDING HIGHER TERMITE TABLE OF CONTENTS MATERIALS AND METHODS 2 • Glycoside hydrolase catalytic domains and carbohydrate binding modules used in searches that are not represented by Pfam HMMs 5 SUPPLEMENTARY TABLES • Table S1. Non-parametric diversity estimators 8 • Table S2. Estimates of gross community structure based on sequence composition binning, and conserved single copy gene phylogenies 8 • Table S3. Summary of numbers glycosyl hydrolases (GHs) and carbon-binding modules (CBMs) discovered in the P3 luminal microbiota 9 • Table S4. Summary of glycosyl hydrolases, their binning information, and activity screening results 13 • Table S5. Comparison of abundance of glycosyl hydrolases in different single organism genomes and metagenome datasets 17 • Table S6. Comparison of abundance of glycosyl hydrolases in different single organism genomes (continued) 20 • Table S7. Phylogenetic characterization of the termite gut metagenome sequence dataset, based on compositional phylogenetic analysis 23 • Table S8. Counts of genes classified to COGs corresponding to different hydrogenase families 24 • Table S9. Fe-only hydrogenases (COG4624, large subunit, C-terminal domain) identified in the P3 luminal microbiota. 25 • Table S10. Gene clusters overrepresented in termite P3 luminal microbiota versus soil, ocean and human gut metagenome datasets. 29 • Table S11. Operational taxonomic unit (OTU) representatives of 16S rRNA sequences obtained from the P3 luminal fluid of Nasutitermes spp. 30 SUPPLEMENTARY FIGURES • Fig. S1. Phylogenetic identification of termite host species 38 • Fig. S2. Accumulation curves of 16S rRNA genes obtained from the P3 luminal microbiota 39 • Fig. S3. Phylogenetic diversity of P3 luminal microbiota within the phylum Spirocheates 40 • Fig. -

United States Patent (19) 11 Patent Number: 5,981,835 Austin-Phillips Et Al
USOO598.1835A United States Patent (19) 11 Patent Number: 5,981,835 Austin-Phillips et al. (45) Date of Patent: Nov. 9, 1999 54) TRANSGENIC PLANTS AS AN Brown and Atanassov (1985), Role of genetic background in ALTERNATIVE SOURCE OF Somatic embryogenesis in Medicago. Plant Cell Tissue LIGNOCELLULOSC-DEGRADING Organ Culture 4:107-114. ENZYMES Carrer et al. (1993), Kanamycin resistance as a Selectable marker for plastid transformation in tobacco. Mol. Gen. 75 Inventors: Sandra Austin-Phillips; Richard R. Genet. 241:49-56. Burgess, both of Madison; Thomas L. Castillo et al. (1994), Rapid production of fertile transgenic German, Hollandale; Thomas plants of Rye. Bio/Technology 12:1366–1371. Ziegelhoffer, Madison, all of Wis. Comai et al. (1990), Novel and useful properties of a chimeric plant promoter combining CaMV 35S and MAS 73 Assignee: Wisconsin Alumni Research elements. Plant Mol. Biol. 15:373-381. Foundation, Madison, Wis. Coughlan, M.P. (1988), Staining Techniques for the Detec tion of the Individual Components of Cellulolytic Enzyme 21 Appl. No.: 08/883,495 Systems. Methods in Enzymology 160:135-144. de Castro Silva Filho et al. (1996), Mitochondrial and 22 Filed: Jun. 26, 1997 chloroplast targeting Sequences in tandem modify protein import specificity in plant organelles. Plant Mol. Biol. Related U.S. Application Data 30:769-78O. 60 Provisional application No. 60/028,718, Oct. 17, 1996. Divne et al. (1994), The three-dimensional crystal structure 51 Int. Cl. ............................. C12N 15/82; C12N 5/04; of the catalytic core of cellobiohydrolase I from Tricho AO1H 5/00 derma reesei. Science 265:524-528. -

Review Article Pullulanase: Role in Starch Hydrolysis and Potential Industrial Applications
Hindawi Publishing Corporation Enzyme Research Volume 2012, Article ID 921362, 14 pages doi:10.1155/2012/921362 Review Article Pullulanase: Role in Starch Hydrolysis and Potential Industrial Applications Siew Ling Hii,1 Joo Shun Tan,2 Tau Chuan Ling,3 and Arbakariya Bin Ariff4 1 Department of Chemical Engineering, Faculty of Engineering and Science, Universiti Tunku Abdul Rahman, 53300 Kuala Lumpur, Malaysia 2 Institute of Bioscience, Universiti Putra Malaysia, 43400 Serdang, Selangor, Malaysia 3 Institute of Biological Sciences, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia 4 Department of Bioprocess Technology, Faculty of Biotechnology and Biomolecular Sciences, Universiti Putra Malaysia, 43400 Serdang, Selangor, Malaysia Correspondence should be addressed to Arbakariya Bin Ariff, [email protected] Received 26 March 2012; Revised 12 June 2012; Accepted 12 June 2012 Academic Editor: Joaquim Cabral Copyright © 2012 Siew Ling Hii et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The use of pullulanase (EC 3.2.1.41) has recently been the subject of increased applications in starch-based industries especially those aimed for glucose production. Pullulanase, an important debranching enzyme, has been widely utilised to hydrolyse the α-1,6 glucosidic linkages in starch, amylopectin, pullulan, and related oligosaccharides, which enables a complete and efficient conversion of the branched polysaccharides into small fermentable sugars during saccharification process. The industrial manufacturing of glucose involves two successive enzymatic steps: liquefaction, carried out after gelatinisation by the action of α- amylase; saccharification, which results in further transformation of maltodextrins into glucose. -

GRAS Notice 000099: Pullulan
United States Department of Agriculture Agricultural Marketing Service | National Organic Program Document Cover Sheet https://www.ams.usda.gov/rules-regulations/organic/national-list/petitioned Document Type: ☒ National List Petition or Petition Update A petition is a request to amend the USDA National Organic Program’s National List of Allowed and Prohibited Substances (National List). Any person may submit a petition to have a substance evaluated by the National Organic Standards Board (7 CFR 205.607(a)). Guidelines for submitting a petition are available in the NOP Handbook as NOP 3011, National List Petition Guidelines. Petitions are posted for the public on the NOP website for Petitioned Substances. ☐ Technical Report A technical report is developed in response to a petition to amend the National List. Reports are also developed to assist in the review of substances that are already on the National List. Technical reports are completed by third-party contractors and are available to the public on the NOP website for Petitioned Substances. Contractor names and dates completed are available in the report. January 31, 2018 National List Manager USDA/AMS/NOP, Standards Division 1400 Independence Ave. SW Room 2648-So., Ag Stop 0268 Washington, DC 20250-0268 RE: Petition to add Pullulan to the National List at §205.605(a) as an allowed nonsynthetic ingredient in tablets and capsules for dietary supplements labeled “made with organic (specified ingredients or food group(s)).” Dear National List Manager: The Organic Trade Association1 is -

Genomic and Transcriptomic Analysis of Carbohydrate Utilization by Paenibacillus Sp
Sawhney et al. BMC Genomics (2016) 17:131 DOI 10.1186/s12864-016-2436-5 RESEARCH ARTICLE Open Access Genomic and transcriptomic analysis of carbohydrate utilization by Paenibacillus sp. JDR-2: systems for bioprocessing plant polysaccharides Neha Sawhney1†, Casey Crooks2, Virginia Chow1, James F. Preston1* and Franz J. St John2*† Abstract Background: Polysaccharides comprising plant biomass are potential resources for conversion to fuels and chemicals. These polysaccharides include xylans derived from the hemicellulose of hardwoods and grasses, soluble β-glucans from cereals and starch as the primary form of energy storage in plants. Paenibacillus sp. JDR-2 (Pjdr2) has evolved a system for bioprocessing xylans. The central component of this xylan utilization system is a multimodular glycoside hydrolase family 10 (GH10) endoxylanase with carbohydrate binding modules (CBM) for binding xylans and surface layer homology (SLH) domains for cell surface anchoring. These attributes allow efficient utilization of xylans by generating oligosaccharides proximal to the cell surface for rapid assimilation. Coordinate expression of genes in response to growth on xylans has identified regulons contributing to depolymerization, importation of oligosaccharides and intracellular processing to generate xylose as well as arabinose and methylglucuronate. The genome of Pjdr2 encodes several other putative surface anchored multimodular enzymes including those for utilization of β-1,3/1,4 mixed linkage soluble glucan and starch. Results: To further define polysaccharide utilization systems in Pjdr2, its transcriptome has been determined by RNA sequencing following growth on barley-derived soluble β-glucan, starch, cellobiose, maltose, glucose, xylose and arabinose. The putative function of genes encoding transcriptional regulators, ABC transporters, and glycoside hydrolases belonging to the corresponding substrate responsive regulon were deduced by their coordinate expression and locations in the genome. -

WO 2015/130881 Al 3 September 2015 (03.09.2015) P O P CT
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/130881 Al 3 September 2015 (03.09.2015) P O P CT (51) International Patent Classification: (72) Inventors: NAGY, Kevin D.; 106 Steven Lane, Wilming C12P 19/16 (2006.01) C12P 19/12 (2006.01) ton, Delaware 19808 (US). HAGO, Erwin Columbus; CUP 19/18 (2006.01) C12P 19/08 (2006.01) 2901 6th Street Sw Apt. 24, Cedar Rapids, Iowa 52404 CUP 19/04 (2006.01) (US). SHETTY, Jayarama K.; 4806 Braxton Place, Pleasonton, California 94566 (US). HENNESSEY, Susan (21) International Application Number: Marie; 32 Truman Lane, Avondale, Pennsylvania 193 11 PCT/US20 15/0 17644 (US). DICOSIMO, Robert; 1607 Masters Way, Chadds (22) International Filing Date: Ford, Pennsylvania 193 17-9720 (US). HUA, Ling; 126 26 February 2015 (26.02.2015) Hockessin Drive, Hockessin, Delaware 19707 (US). RAMIREZ, Rodrigo; Rua Alfredo Ribeiro Nogueira, 280. (25) Filing Language: English Casa08, Campinas, 13092-480 Sao Paulo (BR). TANG, Publication Language: English Zhongmei; Room 602, Building 100, Lane 1100, Gudai Road, Shanghai 201 102 (CN). YU, Zheyong; Room 501, Priority Data: Building 17, Lane 150, Guangyue Road, Hongkou District, 61/945,233 27 February 2014 (27.02.2014) US Shanghai 200000 (CN). 61/945,241 27 February 2014 (27.02.2014) US 62/004,300 29 May 2014 (29.05.2014) us (74) Agent: CHESIRE, Dennis; E. I. du Pont de Nemours and 62/004,305 29 May 2014 (29.05.2014) us Company, Legal Patent Records Center, Chestnut Run 62/004,308 29 May 2014 (29.05.2014) us Plaza 721/2640, 974 Centre Road, PO Box 291 5 Wilming 62/004,290 29 May 2014 (29.05.2014) us ton, Delaware 19805 (US). -

(12) United States Patent (10) Patent No.: US 6,468,955 B1 Smets Et Al
USOO6468955B1 (12) United States Patent (10) Patent No.: US 6,468,955 B1 Smets et al. (45) Date of Patent: Oct. 22, 2002 (54) LAUNDRY DETERGENT AND/OR FABRIC FOREIGN PATENT DOCUMENTS CARE COMPOSITIONS COMPRISINGA MODIFIED ENZYME WO WO 94/07998 4/1994 ............ C12N/9/42 WO WO 94/29460 12/1994 ... C12N/15/62 (75) Inventors: Johan Smets, Lubbeek (BE); Jean-Luc WO WO 97/07203 2/1997 C12N/11/12 Philippe Bettiol, Brussels (BE); WO WO 97/24421 7/1997 WO WO-97/24421 * 7/1997 ........... C11D/3/386 Stanton Lane Boyer, Fairfield, OH WO WO-97/28243 * 8/1997 ..... ... C11D/3/386 (US); Alfred Busch, Londerzeel (BE) WO WO 97/28256 8/1997 ..... ... C12N/9/00 WO WO 97/301.48 8/1997 ..... ... C12N/9/96 (73) Assignee: The Proctor & Gamble Company, WO WO 98/OO500 1/1998 ..... ... C11D/3/386 Cincinnati, OH (US) WO WO 98/16633 4/1998 ..... ... C12N/9/26 (*) Notice: Subject to any disclaimer, the term of this WO WO 98/284.11 7/1998 ............ C12N/9/42 patent is extended or adjusted under 35 U.S.C. 154(b) by 0 days. OTHER PUBLICATIONS (21) Appl. No.: 09/674,478 Characterization of a Double Cellulose-Binding Domain No (22) PCT Filed: Apr. 30, 1999 date. (86) PCT No.: PCT/US99/09453 * cited by examiner S371 (c)(1), (2), (4) Date: Nov. 1, 2000 Primary Examiner Mark Kopec (87) PCT Pub. No.: WO99/57252 ASSistant Examiner-Eisa Elhilo (74) Attorney, Agent, or Firm-Frank Taffy; C. Brant Cook; PCT Pub. Date: Nov. 11, 1999 K. -

(12) United States Patent (10) Patent No.: US 9,689,046 B2 Mayall Et Al
USOO9689046B2 (12) United States Patent (10) Patent No.: US 9,689,046 B2 Mayall et al. (45) Date of Patent: Jun. 27, 2017 (54) SYSTEM AND METHODS FOR THE FOREIGN PATENT DOCUMENTS DETECTION OF MULTIPLE CHEMICAL WO O125472 A1 4/2001 COMPOUNDS WO O169245 A2 9, 2001 (71) Applicants: Robert Matthew Mayall, Calgary (CA); Emily Candice Hicks, Calgary OTHER PUBLICATIONS (CA); Margaret Mary-Flora Bebeselea, A. et al., “Electrochemical Degradation and Determina Renaud-Young, Calgary (CA); David tion of 4-Nitrophenol Using Multiple Pulsed Amperometry at Christopher Lloyd, Calgary (CA); Lisa Graphite Based Electrodes', Chem. Bull. “Politehnica” Univ. Kara Oberding, Calgary (CA); Iain (Timisoara), vol. 53(67), 1-2, 2008. Fraser Scotney George, Calgary (CA) Ben-Yoav. H. et al., “A whole cell electrochemical biosensor for water genotoxicity bio-detection”. Electrochimica Acta, 2009, 54(25), 6113-6118. (72) Inventors: Robert Matthew Mayall, Calgary Biran, I. et al., “On-line monitoring of gene expression'. Microbi (CA); Emily Candice Hicks, Calgary ology (Reading, England), 1999, 145 (Pt 8), 2129-2133. (CA); Margaret Mary-Flora Da Silva, P.S. et al., “Electrochemical Behavior of Hydroquinone Renaud-Young, Calgary (CA); David and Catechol at a Silsesquioxane-Modified Carbon Paste Elec trode'. J. Braz. Chem. Soc., vol. 24, No. 4, 695-699, 2013. Christopher Lloyd, Calgary (CA); Lisa Enache, T. A. & Oliveira-Brett, A. M., "Phenol and Para-Substituted Kara Oberding, Calgary (CA); Iain Phenols Electrochemical Oxidation Pathways”, Journal of Fraser Scotney George, Calgary (CA) Electroanalytical Chemistry, 2011, 1-35. Etesami, M. et al., “Electrooxidation of hydroquinone on simply prepared Au-Pt bimetallic nanoparticles'. Science China, Chem (73) Assignee: FREDSENSE TECHNOLOGIES istry, vol. -
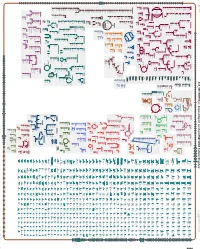
Generate Metabolic Map Poster
Authors: Pallavi Subhraveti Ron Caspi Quang Ong Peter D Karp An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Ingrid Keseler Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_900114035Cyc: Amycolatopsis sacchari DSM 44468 Cellular Overview Connections between pathways are omitted for legibility. -

Potential and Utilization of Thermophiles and Thermostable Enzymes in Biorefining Pernilla Turner, Gashaw Mamo and Eva Nordberg Karlsson*
Microbial Cell Factories BioMed Central Review Open Access Potential and utilization of thermophiles and thermostable enzymes in biorefining Pernilla Turner, Gashaw Mamo and Eva Nordberg Karlsson* Address: Dept Biotechnology, Center for Chemistry and Chemical Engineering, Lund University, P.O. Box 124, SE-221 00 Lund, Sweden Email: Pernilla Turner - [email protected]; Gashaw Mamo - [email protected]; Eva Nordberg Karlsson* - [email protected] * Corresponding author Published: 15 March 2007 Received: 4 January 2007 Accepted: 15 March 2007 Microbial Cell Factories 2007, 6:9 doi:10.1186/1475-2859-6-9 This article is available from: http://www.microbialcellfactories.com/content/6/1/9 © 2007 Turner et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Abstract In today's world, there is an increasing trend towards the use of renewable, cheap and readily available biomass in the production of a wide variety of fine and bulk chemicals in different biorefineries. Biorefineries utilize the activities of microbial cells and their enzymes to convert biomass into target products. Many of these processes require enzymes which are operationally stable at high temperature thus allowing e.g. easy mixing, better substrate solubility, high mass transfer rate, and lowered risk of contamination. Thermophiles have often been proposed as sources of industrially relevant thermostable enzymes. Here we discuss existing and potential applications of thermophiles and thermostable enzymes with focus on conversion of carbohydrate containing raw materials. -

Long-Term Warming in a Mediterranean-Type Grassland Affects Soil Bacterial Functional Potential but Not Bacterial Taxonomic Composition
www.nature.com/npjbiofilms ARTICLE OPEN Long-term warming in a Mediterranean-type grassland affects soil bacterial functional potential but not bacterial taxonomic composition Ying Gao1,2, Junjun Ding2,3, Mengting Yuan4, Nona Chiariello5, Kathryn Docherty6, Chris Field 5, Qun Gao2, Baohua Gu 7, Jessica Gutknecht8,9, Bruce A. Hungate 10,11, Xavier Le Roux12, Audrey Niboyet13,14,QiQi2, Zhou Shi4, Jizhong Zhou2,4,15 and ✉ Yunfeng Yang2 Climate warming is known to impact ecosystem composition and functioning. However, it remains largely unclear how soil microbial communities respond to long-term, moderate warming. In this study, we used Illumina sequencing and microarrays (GeoChip 5.0) to analyze taxonomic and functional gene compositions of the soil microbial community after 14 years of warming (at 0.8–1.0 °C for 10 years and then 1.5–2.0 °C for 4 years) in a Californian grassland. Long-term warming had no detectable effect on the taxonomic composition of soil bacterial community, nor on any plant or abiotic soil variables. In contrast, functional gene compositions differed between warming and control for bacterial, archaeal, and fungal communities. Functional genes associated with labile carbon (C) degradation increased in relative abundance in the warming treatment, whereas those associated with recalcitrant C degradation decreased. A number of functional genes associated with nitrogen (N) cycling (e.g., denitrifying genes encoding nitrate-, nitrite-, and nitrous oxidereductases) decreased, whereas nifH gene encoding nitrogenase increased in the 1234567890():,; warming treatment. These results suggest that microbial functional potentials are more sensitive to long-term moderate warming than the taxonomic composition of microbial community. -

Review Article Pullulanase: Role in Starch Hydrolysis and Potential Industrial Applications
Hindawi Publishing Corporation Enzyme Research Volume 2012, Article ID 921362, 14 pages doi:10.1155/2012/921362 Review Article Pullulanase: Role in Starch Hydrolysis and Potential Industrial Applications Siew Ling Hii,1 Joo Shun Tan,2 Tau Chuan Ling,3 and Arbakariya Bin Ariff4 1 Department of Chemical Engineering, Faculty of Engineering and Science, Universiti Tunku Abdul Rahman, 53300 Kuala Lumpur, Malaysia 2 Institute of Bioscience, Universiti Putra Malaysia, 43400 Serdang, Selangor, Malaysia 3 Institute of Biological Sciences, Faculty of Science, University of Malaya, 50603 Kuala Lumpur, Malaysia 4 Department of Bioprocess Technology, Faculty of Biotechnology and Biomolecular Sciences, Universiti Putra Malaysia, 43400 Serdang, Selangor, Malaysia Correspondence should be addressed to Arbakariya Bin Ariff, [email protected] Received 26 March 2012; Revised 12 June 2012; Accepted 12 June 2012 Academic Editor: Joaquim Cabral Copyright © 2012 Siew Ling Hii et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. The use of pullulanase (EC 3.2.1.41) has recently been the subject of increased applications in starch-based industries especially those aimed for glucose production. Pullulanase, an important debranching enzyme, has been widely utilised to hydrolyse the α-1,6 glucosidic linkages in starch, amylopectin, pullulan, and related oligosaccharides, which enables a complete and efficient conversion of the branched polysaccharides into small fermentable sugars during saccharification process. The industrial manufacturing of glucose involves two successive enzymatic steps: liquefaction, carried out after gelatinisation by the action of α- amylase; saccharification, which results in further transformation of maltodextrins into glucose.