Blueprint Genetics Metaphyseal Dysplasia Panel
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The National Economic Burden of Rare Disease Study February 2021
Acknowledgements This study was sponsored by the EveryLife Foundation for Rare Diseases and made possible through the collaborative efforts of the national rare disease community and key stakeholders. The EveryLife Foundation thanks all those who shared their expertise and insights to provide invaluable input to the study including: the Lewin Group, the EveryLife Community Congress membership, the Technical Advisory Group for this study, leadership from the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH), the Undiagnosed Diseases Network (UDN), the Little Hercules Foundation, the Rare Disease Legislative Advocates (RDLA) Advisory Committee, SmithSolve, and our study funders. Most especially, we thank the members of our rare disease patient and caregiver community who participated in this effort and have helped to transform their lived experience into quantifiable data. LEWIN GROUP PROJECT STAFF Grace Yang, MPA, MA, Vice President Inna Cintina, PhD, Senior Consultant Matt Zhou, BS, Research Consultant Daniel Emont, MPH, Research Consultant Janice Lin, BS, Consultant Samuel Kallman, BA, BS, Research Consultant EVERYLIFE FOUNDATION PROJECT STAFF Annie Kennedy, BS, Chief of Policy and Advocacy Julia Jenkins, BA, Executive Director Jamie Sullivan, MPH, Director of Policy TECHNICAL ADVISORY GROUP Annie Kennedy, BS, Chief of Policy & Advocacy, EveryLife Foundation for Rare Diseases Anne Pariser, MD, Director, Office of Rare Diseases Research, National Center for Advancing Translational Sciences (NCATS), National Institutes of Health Elisabeth M. Oehrlein, PhD, MS, Senior Director, Research and Programs, National Health Council Christina Hartman, Senior Director of Advocacy, The Assistance Fund Kathleen Stratton, National Academies of Science, Engineering and Medicine (NASEM) Steve Silvestri, Director, Government Affairs, Neurocrine Biosciences Inc. -

Increased Nuchal Translucency Precision Panel
Increased Nuchal Translucency Precision Panel Overview Increased Nuchal Translucency (NT) is defined as an abnormal accumulation of fluid in the nuchal area, which is visualized as a thickened sonolucent area. It is a standardized measure obtained between 11 and 14 weeks of gestation to calculate the risk of a fetus being affected by a chromosomal aneuploidy. NT>3.5mm has been found to be associated with fetal chromosomal abnormalities and single-gene disorders as well as cardiac defects and other structural abnormalities in euploid and aneuploid fetuses. Proportionally as NT increases, even with a normal karyotype, there is a higher risk of adverse pregnancy outcomes such as miscarriage, intrauterine death, congenital heart defects and numerous other structural and genetic syndromes. There is not one single cause of increased NT, it is based on a complex and multifactorial process, linked to one or more embryonic processes. It has been shown that a persistently increased NT with a normal karyotype and aCGH has a 4-10% probability of being associated to Noonan Syndrome and/or other RASopathies using Whole Exome Sequencing (WES). However, the general tendency following detection of isolated enlarged NT in an euploid fetus is that most babies with normal detailed ultrasound examination and echocardiography will have uneventful outcomes. The Igenomix Increased Nuchal Translucency Precision Panel can be used to make a directed and accurate prenatal differential diagnosis of increased nuchal translucency in patients with or without a normal karyotype ultimately leading to a better management and prognosis of the associated comorbidities. It provides a comprehensive analysis of the genes involved in this disease using next-generation sequencing (NGS) to fully understand the spectrum of relevant genes involved. -
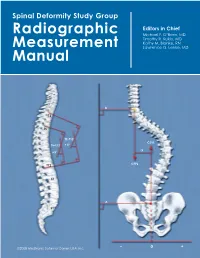
Spinal Deformity Study Group
Spinal Deformity Study Group Editors in Chief Radiographic Michael F. O’Brien, MD Timothy R. Kuklo, MD Kathy M. Blanke, RN Measurement Lawrence G. Lenke, MD Manual B T2 T5 T2–T12 CSVL T5–T12 +X° -X +X° C7PL T12 L2 A S1 ©2008 Medtronic Sofamor Danek USA, Inc. – 0 + Radiographic Measurement Manual Editors in Chief Michael F. O’Brien, MD Timothy R. Kuklo, MD Kathy M. Blanke, RN Lawrence G. Lenke, MD Section Editors Keith H. Bridwell, MD Kathy M. Blanke, RN Christopher L. Hamill, MD William C. Horton, MD Timothy R. Kuklo, MD Hubert B. Labelle, MD Lawrence G. Lenke, MD Michael F. O’Brien, MD David W. Polly Jr, MD B. Stephens Richards III, MD Pierre Roussouly, MD James O. Sanders, MD ©2008 Medtronic Sofamor Danek USA, Inc. Acknowledgements Radiographic Measurement Manual The radiographic measurement manual has been developed to present standardized techniques for radiographic measurement. In addition, this manual will serve as a complimentary guide for the Spinal Deformity Study Group’s radiographic measurement software. Special thanks to the following members of the Spinal Deformity Study Group in the development of this manual. Sigurd Berven, MD Hubert B. Labelle, MD Randal Betz, MD Lawrence G. Lenke, MD Fabien D. Bitan, MD Thomas G. Lowe, MD John T. Braun, MD John P. Lubicky, MD Keith H. Bridwell, MD Steven M. Mardjetko, MD Courtney W. Brown, MD Richard E. McCarthy, MD Daniel H. Chopin, MD Andrew A. Merola, MD Edgar G. Dawson, MD Michael Neuwirth, MD Christopher DeWald, MD Peter O. Newton, MD Mohammad Diab, MD Michael F. -

Lordosis, Kyphosis, and Scoliosis
SPINAL CURVATURES: LORDOSIS, KYPHOSIS, AND SCOLIOSIS The human spine normally curves to aid in stability or balance and to assist in absorbing shock during movement. These gentle curves can be seen from the side or lateral view of the spine. When viewed from the back, the spine should run straight down the middle of the back. When there are abnormalities or changes in the natural spinal curvature, these abnormalities are named with the following conditions and include the following symptoms. LORDOSIS Some lordosis is normal in the lower portion or, lumbar section, of the human spine. A decreased or exaggerated amount of lordosis that is causing spinal instability is a condition that may affect some patients. Symptoms of Lordosis include: ● Appearance of sway back where the lower back region has a pronounced curve and looks hollow with a pronounced buttock area ● Difficulty with movement in certain directions ● Low back pain KYPHOSIS This condition is diagnosed when the patient has a rounded upper back and the spine is bent over or curved more than 50 degrees. Symptoms of Kyphosis include: ● Curved or hunched upper back ● Patient’s head that leans forward ● May have upper back pain ● Experiences upper back discomfort after movement or exercise SCOLIOSIS The most common of the three curvatures. This condition is diagnosed when the spine looks like a “s” or “c” from the back. The spine is not straight up and down but has a curve or two running side-to-side. Sagittal Balance Definition • Sagittal= front-to-back direction (sagittal plane) • Imbalance= Lack of harmony or balance Etiology • Excessive lordosis (backwards lean) or kyphosis (forward lean) • Traumatic injury • Previous spinal fusion that disrupted sagittal balance Effects • Low back pain • Difficulty walking • Inability to look straight ahead when upright The most ergonomic and natural posture is to maintain neutral balance, with the head positioned over the shoulders and pelvis. -

Marble Bone Disease: a Rare Bone Disorder
Open Access Case Report DOI: 10.7759/cureus.339 Marble Bone Disease: A Rare Bone Disorder Eswaran Arumugam 1 , Maheswari Harinathbabu 2 , Ranjani Thillaigovindan 1 , Geetha Prabhu 1 1. Prosthodontics, Thai Moogambigai Dental College and Hospital 2. Oral Medicine and Radiology, Siva Multi Speciality Dental Clinic Corresponding author: Eswaran Arumugam, [email protected] Abstract Osteopetrosis, or marble bone disease, is a rare skeletal disorder due to a defective function of the osteoclasts. This defect renders bones more susceptible to osteomyelitis due to decreased vascularity. This disorder is inherited as autosomal dominant and autosomal recessive. Healthcare professionals should urge these patients to maintain their oral health as well as general health, as this condition makes these patients more susceptible to frequent infections and fractures. This case report emphasizes the signs and symptoms of marble bone disease and presents clinical and radiographic findings. Categories: Physical Medicine & Rehabilitation, Miscellaneous Keywords: osteopetrosis, marble bone disease, autosomal recessive, dense sclerotic bone Introduction Osteopetrosis (literally "stone bone," also known as marble bone disease or Albers-Schonberg disease) is an extremely rare inherited disorder where the bones harden and become denser. The disorder can cause osteosclerosis. The estimated prevalence of osteopetrosis is 1 in 100,000 to 500,000. It presents in two major clinical forms-a benign autosomal dominant form and a malignant autosomal recessive form. The autosomal dominant adult (benign) form is associated with few, if any, symptoms, and the autosomal recessive infantile (malignant) form is typically fatal during infancy or early childhood if untreated [1]. A rarer autosomal recessive (intermediate) form presents during childhood with some signs and symptoms of malignant osteopetrosis. -

Supporting Information
Supporting Information Torkamani et al. 10.1073/pnas.0802403105 Materials and Methods kinase sequences used to generate conserved motifs, as in Kannan Kinase Identifiers. Kinase protein and DNA reference sequences et al. (3), the Gibbs motif sampling method identifies characteristic were obtained from Kinbase. These reference sequences were motifs for each individual subdomain of the kinase catalytic core, used as the basis to assign various gene identifiers (including which are then used to generate high confidence motif-based Ensembl gene IDs, HGNC gene symbols, and Entrez gene IDs) Markov chain Monte Carlo multiple alignments based upon these to every known human protein kinase. Ultimately, only eukary- motifs (4). These subdomains compromise the core structural otic protein kinases, that is, all human protein kinases except components of the protein kinase catalytic core. Intervening re- those belonging to the atypical protein kinase family, were gions between these subdomains were not aligned. considered in this study. The various gene identifiers were assigned as follows: Ensembl Mapping to Multiple Alignments and Generation of Logo Figures. A Gene ID’s were determined for each protein kinase by BLAST- nonredundant set of SNPs was generated to be mapped to the ing the reference Kinbase protein sequence against the Ensembl alignment computationally. That is, if multiple disease or com- database (www.ensembl.org/Homo sapiens/blastview). The En- mon SNPs have been observed at a particular position within a sembl Gene ID of the top hit was assigned to the protein kinase. particular protein kinase, it is only considered once in our The Ensembl Gene ID was then used as a query in Biomart analysis. -

Metaphyseal Dysplasia: a Rare Case Report Dildip Khanal* Karuna Foundation Nepal “Saving Children from Disability, One by One”, Nepal
ical C lin as Khanal, J Clin Case Rep 2016, 6:2 C e f R o l e a p DOI: 10.4172/2165-7920.1000726 n o r r t u s o J Journal of Clinical Case Reports ISSN: 2165-7920 Case Report Open Access Metaphyseal Dysplasia: A Rare Case Report Dildip Khanal* Karuna Foundation Nepal “Saving Children from Disability, One by One”, Nepal Abstract Metaphyseal dysplasia is a very rare inherited bone disorder. Here is a case report and possible treatment options for 11 years old child, detected by Karuna foundation Nepal. Keywords: Metaphyseal dysplasia; Pyle; Therapeutic rehabilitation; Karuna foundation Nepal Background Metaphyseal dysplasia also known as Pyle disease is a heterogeneous group of disorders, characterized by the metaphyseal changes of the tubular bones with normal epiphyses. The disease was described briefly by Pyle in 1931 [1,2]. Incidence occurs at a rate of two to three newborns per 10,000 births involving the proliferative and hypertrophic zone of Figure 1: Bilateral genu varum deformity. the physis (epiphysis is normal). Jansen, Schmid and McKusick are the three sub-types with a few reports worldwide [3-9]. Karuna foundation Nepal (KFN) is a non-governmental organization which believes in a world in which each individual, with or without disabilities, has equal access to good quality health care, can lead a dignified life, and can participate as much as possible in community life. KFN approach is entrepreneurial and action oriented, working towards setting up and strengthening existing local health care system, stimulating community participation and responsibility- including health promotion, prevention and rehabilitation through Figure 2: Flat foot. -

Repercussions of Inborn Errors of Immunity on Growth☆ Jornal De Pediatria, Vol
Jornal de Pediatria ISSN: 0021-7557 ISSN: 1678-4782 Sociedade Brasileira de Pediatria Goudouris, Ekaterini Simões; Segundo, Gesmar Rodrigues Silva; Poli, Cecilia Repercussions of inborn errors of immunity on growth☆ Jornal de Pediatria, vol. 95, no. 1, Suppl., 2019, pp. S49-S58 Sociedade Brasileira de Pediatria DOI: https://doi.org/10.1016/j.jped.2018.11.006 Available in: https://www.redalyc.org/articulo.oa?id=399759353007 How to cite Complete issue Scientific Information System Redalyc More information about this article Network of Scientific Journals from Latin America and the Caribbean, Spain and Journal's webpage in redalyc.org Portugal Project academic non-profit, developed under the open access initiative J Pediatr (Rio J). 2019;95(S1):S49---S58 www.jped.com.br REVIEW ARTICLE ଝ Repercussions of inborn errors of immunity on growth a,b,∗ c,d e Ekaterini Simões Goudouris , Gesmar Rodrigues Silva Segundo , Cecilia Poli a Universidade Federal do Rio de Janeiro (UFRJ), Faculdade de Medicina, Departamento de Pediatria, Rio de Janeiro, RJ, Brazil b Universidade Federal do Rio de Janeiro (UFRJ), Instituto de Puericultura e Pediatria Martagão Gesteira (IPPMG), Curso de Especializac¸ão em Alergia e Imunologia Clínica, Rio de Janeiro, RJ, Brazil c Universidade Federal de Uberlândia (UFU), Faculdade de Medicina, Departamento de Pediatria, Uberlândia, MG, Brazil d Universidade Federal de Uberlândia (UFU), Hospital das Clínicas, Programa de Residência Médica em Alergia e Imunologia Pediátrica, Uberlândia, MG, Brazil e Universidad del Desarrollo, -

Skeletal Dysplasias
Skeletal Dysplasias North Carolina Ultrasound Society Keisha L.B. Reddick, MD Wilmington Maternal Fetal Medicine Development of the Skeleton • 6 weeks – vertebrae • 7 weeks – skull • 8 wk – clavicle and mandible – Hyaline cartilage • Ossification – 7-12 wk – diaphysis appears – 12-16 wk metacarpals and metatarsals – 20+ wk pubis, calus, calcaneus • Visualization of epiphyseal ossification centers Epidemiology • Overall 9.1 per 1000 • Lethal 1.1 per 10,000 – Thanatophoric 1/40,000 – Osteogenesis Imperfecta 0.18 /10,000 – Campomelic 0.1 /0,000 – Achondrogenesis 0.1 /10,000 • Non-lethal – Achondroplasia 15 in 10,000 Most Common Skeletal Dysplasia • Thantophoric dysplasia 29% • Achondroplasia 15% • Osteogenesis imperfecta 14% • Achondrogenesis 9% • Campomelic dysplasia 2% Definition/Terms • Rhizomelia – proximal segment • Mezomelia –intermediate segment • Acromelia – distal segment • Micromelia – all segments • Campomelia – bowing of long bones • Preaxial – radial/thumb or tibial side • Postaxial – ulnar/little finger or fibular Long Bone Segments Counseling • Serial ultrasound • Genetic counseling • Genetic testing – Amniocentesis • Postnatal – Delivery center – Radiographs Assessment • Which segment is affected • Assessment of distal extremities • Any curvatures, fracture or clubbing noted • Are metaphyseal changes present • Hypoplastic or absent bones • Assessment of the spinal canal • Assessment of thorax. Skeletal Dysplasia Lethal Non-lethal • Thanatophoric • Achondroplasia • OI type II • OI type I, III, IV • Achondrogenesis • Hypochondroplasia -

Inherited Renal Tubulopathies—Challenges and Controversies
G C A T T A C G G C A T genes Review Inherited Renal Tubulopathies—Challenges and Controversies Daniela Iancu 1,* and Emma Ashton 2 1 UCL-Centre for Nephrology, Royal Free Campus, University College London, Rowland Hill Street, London NW3 2PF, UK 2 Rare & Inherited Disease Laboratory, London North Genomic Laboratory Hub, Great Ormond Street Hospital for Children National Health Service Foundation Trust, Levels 4-6 Barclay House 37, Queen Square, London WC1N 3BH, UK; [email protected] * Correspondence: [email protected]; Tel.: +44-2381204172; Fax: +44-020-74726476 Received: 11 February 2020; Accepted: 29 February 2020; Published: 5 March 2020 Abstract: Electrolyte homeostasis is maintained by the kidney through a complex transport function mostly performed by specialized proteins distributed along the renal tubules. Pathogenic variants in the genes encoding these proteins impair this function and have consequences on the whole organism. Establishing a genetic diagnosis in patients with renal tubular dysfunction is a challenging task given the genetic and phenotypic heterogeneity, functional characteristics of the genes involved and the number of yet unknown causes. Part of these difficulties can be overcome by gathering large patient cohorts and applying high-throughput sequencing techniques combined with experimental work to prove functional impact. This approach has led to the identification of a number of genes but also generated controversies about proper interpretation of variants. In this article, we will highlight these challenges and controversies. Keywords: inherited tubulopathies; next generation sequencing; genetic heterogeneity; variant classification. 1. Introduction Mutations in genes that encode transporter proteins in the renal tubule alter kidney capacity to maintain homeostasis and cause diseases recognized under the generic name of inherited tubulopathies. -

Current Overview of Osteogenesis Imperfecta
medicina Review Current Overview of Osteogenesis Imperfecta Mari Deguchi *, Shunichiro Tsuji , Daisuke Katsura , Kyoko Kasahara, Fuminori Kimura and Takashi Murakami Department of Obstetrics & Gynecology, Shiga University of Medical Science, Otsu 520-2192, Shiga, Japan; [email protected] (S.T.); [email protected] (D.K.); [email protected] (K.K.); [email protected] (F.K.); [email protected] (T.M.) * Correspondence: [email protected] Abstract: Osteogenesis imperfecta (OI), or brittle bone disease, is a heterogeneous disorder charac- terised by bone fragility, multiple fractures, bone deformity, and short stature. OI is a heterogeneous disorder primarily caused by mutations in the genes involved in the production of type 1 collagen. Severe OI is perinatally lethal, while mild OI can sometimes not be recognised until adulthood. Severe or lethal OI can usually be diagnosed using antenatal ultrasound and confirmed by various imaging modalities and genetic testing. The combination of imaging parameters obtained by ultrasound, computed tomography (CT), and magnetic resource imaging (MRI) can not only detect OI accurately but also predict lethality before birth. Moreover, genetic testing, either noninvasive or invasive, can further confirm the diagnosis prenatally. Early and precise diagnoses provide parents with more time to decide on reproductive options. The currently available postnatal treatments for OI are not curative, and individuals with severe OI suffer multiple fractures and bone deformities throughout their lives. In utero mesenchymal stem cell transplantation has been drawing attention as a promising therapy for severe OI, and a clinical trial to assess the safety and efficacy of cell therapy is currently ongoing. -

Peds Ortho: What Is Normal, What Is Not, and When to Refer
Peds Ortho: What is normal, what is not, and when to refer Future of Pedatrics June 10, 2015 Matthew E. Oetgen Benjamin D. Martin Division of Orthopaedic Surgery AGENDA • Definitions • Lower Extremity Deformity • Spinal Alignment • Back Pain LOWER EXTREMITY ALIGNMENT DEFINITIONS coxa = hip genu = knee cubitus = elbow pes = foot varus valgus “bow-legged” “knock-knee” apex away from midline apex toward midline normal varus hip (coxa vara) varus humerus valgus ankle valgus hip (coxa valga) Genu varum (bow-legged) Genu valgum (knock knee) bow legs and in toeing often together Normal Limb alignment NORMAL < 2 yo physiologic = reassurance, reevaluate @ 2 yo Bow legged 7° knock knee normal Knock knee physiologic = reassurance, reevaluate in future 4 yo abnormal 10 13 yo abnormal + pain 11 Follow-up is essential! 12 Intoeing 1. Femoral anteversion 2. Tibial torsion 3. Metatarsus adductus MOST LIKELY PHYSIOLOGIC AND WILL RESOLVE! BRACES ARE HISTORY! Femoral Anteversion “W” sitters Internal rotation >> External rotation knee caps point in MOST LIKELY PHYSIOLOGIC AND MAY RESOLVE! Internal Tibial Torsion Thigh foot angle MOST LIKELY PHYSIOLOGIC AND WILL RESOLVE BY SCHOOL AGE Foot is rotated inward Internal Tibial Torsion (Fuchs 1996) Metatarsus Adductus • Flexible = correctible • Observe vs. casting CURVED LATERAL BORDER toes point in NOT TO BE CONFUSED WITH… Clubfoot talipes equinovarus adductus internal varus rotation equinus CAN’T DORSIFLEX cavus Clubfoot START19 CASTING JUST AFTER BIRTH Calcaneovalgus Foot • Intrauterine positioning • Resolve