Interleukin-1 Family Cytokines
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Huil36g 169 Data Sheet
Growth Factor Data Sheet GoldBio growth factors are manufactured for RESEARCH USE ONLY and cannot be sold for human consumption! Interleukin-36G (IL36G) is a pro-inflammatory cytokine that plays an important role in the pathophysiology of several diseases. IL36A, IL36B and IL36G; (formerly IL1F6, IL1F8, and IL1F9) are IL1 family members that signal through the IL1 receptor family members IL1Rrp2 (IL1RL2) and IL1RAcP. IL36B is secreted when transfected into 293-T cells and could constitute part of an independent signaling system analogous to that of IL1A and IL1B receptor agonist and interleukin-1 receptor type I (IL1R1). Furthermore, IL36G also can function as an agonist of NFκB activation through the orphan IL1- receptor-related protein 2. Recombinant human IL36G is synthesized as a protein that contains no signal sequence, no prosegment and no potential N-linked glycosylation site.There is a 53% amino acid homology between human and mouse IL36G. IL36G also has a 25-55% amino acid homology with IL36G and IL1RN, IL1B, IL36RN, IL36A, IL37, IL36B and IL1F10. Catalog Number 1110-36E Product Name IL36G (IL-36 gamma), Human (169 a.a.) Recombinant Human Interleukin-36γ IL36G, IL36γ Interleukin 1 Homolog 1 (IL1H1) Interleukin 1-Related Protein 2 (IL1RP2) Interleukin 1 Family, Member 9 (IL1F9) Source Escherichia coli MW 18.7 kDa (169 amino acids) Sequence MRGTPGDADG GGRAVYQSMC KPITGTINDL NQQVWTLQGQ NLVAVPRSDS VTPVTVAVIT CKYPEALEQG RGDPIYLGIQ NPEMCLYCEK VGEQPTLQLK EQKIMDLYGQ PEPVKPFLFY RAKTGRTSTL ESVAFPDWFI ASSKRDQPII LTSELGKSYN TAFELNIND Accession Number Q9NZH8 Purity >95% by SDS-PAGE and HPLC analyses Biological Activity Fully biologically active when compared to standard. The specific activity is determined by its binding ability in a functional ELISA. -

Protein Identities in Evs Isolated from U87-MG GBM Cells As Determined by NG LC-MS/MS
Protein identities in EVs isolated from U87-MG GBM cells as determined by NG LC-MS/MS. No. Accession Description Σ Coverage Σ# Proteins Σ# Unique Peptides Σ# Peptides Σ# PSMs # AAs MW [kDa] calc. pI 1 A8MS94 Putative golgin subfamily A member 2-like protein 5 OS=Homo sapiens PE=5 SV=2 - [GG2L5_HUMAN] 100 1 1 7 88 110 12,03704523 5,681152344 2 P60660 Myosin light polypeptide 6 OS=Homo sapiens GN=MYL6 PE=1 SV=2 - [MYL6_HUMAN] 100 3 5 17 173 151 16,91913397 4,652832031 3 Q6ZYL4 General transcription factor IIH subunit 5 OS=Homo sapiens GN=GTF2H5 PE=1 SV=1 - [TF2H5_HUMAN] 98,59 1 1 4 13 71 8,048185945 4,652832031 4 P60709 Actin, cytoplasmic 1 OS=Homo sapiens GN=ACTB PE=1 SV=1 - [ACTB_HUMAN] 97,6 5 5 35 917 375 41,70973209 5,478027344 5 P13489 Ribonuclease inhibitor OS=Homo sapiens GN=RNH1 PE=1 SV=2 - [RINI_HUMAN] 96,75 1 12 37 173 461 49,94108966 4,817871094 6 P09382 Galectin-1 OS=Homo sapiens GN=LGALS1 PE=1 SV=2 - [LEG1_HUMAN] 96,3 1 7 14 283 135 14,70620005 5,503417969 7 P60174 Triosephosphate isomerase OS=Homo sapiens GN=TPI1 PE=1 SV=3 - [TPIS_HUMAN] 95,1 3 16 25 375 286 30,77169764 5,922363281 8 P04406 Glyceraldehyde-3-phosphate dehydrogenase OS=Homo sapiens GN=GAPDH PE=1 SV=3 - [G3P_HUMAN] 94,63 2 13 31 509 335 36,03039959 8,455566406 9 Q15185 Prostaglandin E synthase 3 OS=Homo sapiens GN=PTGES3 PE=1 SV=1 - [TEBP_HUMAN] 93,13 1 5 12 74 160 18,68541938 4,538574219 10 P09417 Dihydropteridine reductase OS=Homo sapiens GN=QDPR PE=1 SV=2 - [DHPR_HUMAN] 93,03 1 1 17 69 244 25,77302971 7,371582031 11 P01911 HLA class II histocompatibility antigen, -

Interleukin (IL)17A, F and AF in Inflammation: a Study in Collageninduced Arthritis and Rheumatoid Arthritis
bs_bs_banner Clinical and Experimental Immunology ORIGINAL ARTICLE doi:10.1111/cei.12376 Interleukin (IL)-17A, F and AF in inflammation: a study in collagen-induced arthritis and rheumatoid arthritis S. Sarkar,* S. Justa,* M. Brucks,* Summary J. Endres,† D. A. Fox,† X. Zhou,* Interleukin (IL)-17 plays a critical role in inflammation. Most studies to date F. Alnaimat,* B. Whitaker,‡ have elucidated the inflammatory role of IL-17A, often referred to as IL-17. J. C. Wheeler,‡ B. H. Jones§ and IL-17F is a member of the IL-17 family bearing 50% homology to IL-17A S. R. Bommireddy* and can also be present as heterodimer IL-17AF. This study elucidates the *Section of Rheumatology, Department of Medicine, and the Arizona Arthritis Center, distribution and contribution of IL-17A, F and AF in inflammatory arthritis. University of Arizona, Tucson, AZ, †Divison of Neutralizing antibody to IL-17A alone or IL-17F alone or in combination Rheumatology, Department of Internal Medicine, was utilized in the mouse collagen-induced arthritis (CIA) model to eluci- University of Michigan, Ann Arbor, MI, and date the contribution of each subtype in mediating inflammation. IL-17A, F ‡Biologics Research and §Immunology Discovery and AF were all increased during inflammatory arthritis. Neutralization of Research, Janssen Research and Development, IL-17A reduced the severity of arthritis, neutralization of IL-17A+IL-17F had Spring House, PA, USA the same effect as neutralizing IL-17A, while neutralization of IL-17F had no effect. Moreover, significantly higher levels of IL-17A and IL-17F were detected in peripheral blood mononuclear cells (PBMC) from patients with rheumatoid arthritis (RA) in comparison to patients with osteoarthritis (OA). -

Regulation of Osteoblast Differentiation by Cytokine Networks
International Journal of Molecular Sciences Review Regulation of Osteoblast Differentiation by Cytokine Networks Dulshara Sachini Amarasekara 1, Sumi Kim 2 and Jaerang Rho 2,* 1 Department of Zoology and Environment Sciences, Faculty of Science, University of Colombo, Colombo 00300, Sri Lanka; [email protected] 2 Department of Microbiology and Molecular Biology, Chungnam National University, Daejeon 34134, Korea; [email protected] * Correspondence: [email protected]; Tel.: +82-42-821-6420; Fax: +82-42-822-7367 Abstract: Osteoblasts, which are bone-forming cells, play pivotal roles in bone modeling and remod- eling. Osteoblast differentiation, also known as osteoblastogenesis, is orchestrated by transcription factors, such as runt-related transcription factor 1/2, osterix, activating transcription factor 4, special AT-rich sequence-binding protein 2 and activator protein-1. Osteoblastogenesis is regulated by a network of cytokines under physiological and pathophysiological conditions. Osteoblastogenic cytokines, such as interleukin-10 (IL-10), IL-11, IL-18, interferon-γ (IFN-γ), cardiotrophin-1 and oncostatin M, promote osteoblastogenesis, whereas anti-osteoblastogenic cytokines, such as tumor necrosis factor-α (TNF-α), TNF-β, IL-1α, IL-4, IL-7, IL-12, IL-13, IL-23, IFN-α, IFN-β, leukemia inhibitory factor, cardiotrophin-like cytokine, and ciliary neurotrophic factor, downregulate os- teoblastogenesis. Although there are gaps in the body of knowledge regarding the interplay of cytokine networks in osteoblastogenesis, cytokines appear to be potential therapeutic targets in bone- related diseases. Thus, in this study, we review and discuss our osteoblast, osteoblast differentiation, osteoblastogenesis, cytokines, signaling pathway of cytokine networks in osteoblastogenesis. Keywords: osteoblast; osteoblast differentiation; osteoblastogenesis; cytokine; signaling pathway Citation: Amarasekara, D.S.; Kim, S.; Rho, J. -

Interleukin 2 Medical Intensive Care Unit (4MICU)
Interleukin 2 Medical Intensive Care Unit (4MICU) Ronald Reagan UCLA Medical Center 757 Westwood Plaza Los Angeles, CA 90095 Main Phone: (310) 267-7441 Fax: (310) 267-3785 About Our Unit The Medical Intensive Care Unit (MICU) cares Quick for critically ill patients in an intensive care Reference Guide environment, with nursing staff specially trained in the administration of Interleukin 2 therapy. Unit Director / Manager Mark Flitcraft, RN, MSN One registered nurse (RN) is assigned to take (310) 267-9529 care of a maximum of two patients. Our Medical Clinical Nurse Specialist Intensive Care Unit patient rooms are designed Yuhan Kao, RN, MSN, CNS (310) 267-7465 to allow nurses constant visual contact with their patients. As a safety precaution, the Medical Assistant Manager Sherry Xu, RN, BA, CCRN Intensive Care Unit is a closed unit and requires (310) 267-7485 permission to enter by intercom. Clinical Case Manager Each private-patient-care room contains the Connie Lefevre (310) 267-9740 most advanced intensive-care equipment available, including cardiac-monitoring and Clinical Social Worker Codie Lieto emergency-response equipment. The curtains in (310) 267-9741 the room will usually be drawn to keep your room Charge Nurse On-Duty more private. (310) 267-7480 or (310) 267-7482 A brief tour is available on weekdays for patients and visitors interested in walking through the unit Patient Affairs (310) 267-9113 and meeting the staff before arrival. To arrange for a tour, please call the nurse manager at Respiratory Supervisor (310) 267-9529. Orna Molayeme, MA, RCP, RRT, NPS (310) 267-8921 UCLAHEALTH.ORG 1-800-UCLA-MD1 (1-800-825-2631) About Our Unit During Your Stay Quick The Medical Team Reference Guide During each shift, you will be assigned a registered nurse (RN) and a clinical care partner (CCP). -

IL36G Blocking Peptide (CDBP5559) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
IL36G blocking peptide (CDBP5559) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Antigen Description The protein encoded by this gene is a member of the interleukin 1 cytokine family. The activity of this cytokine is mediated by interleukin 1 receptor-like 2 (IL1RL2/IL1R-rp2), and is specifically inhibited by interleukin 1 family, member 5 (IL1F5/IL-1 delta). Interferon-gamma, tumor necrosis factor-alpha and interleukin 1, beta (IL1B) are reported to stimulate the expression of this cytokine in keratinocytes. The expression of this cytokine in keratinocytes can also be induced by a contact hypersensitivity reaction or herpes simplex virus infection. This gene and eight other interleukin 1 family genes form a cytokine gene cluster on chromosome 2. Two alternatively spliced transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Jun 2013] Immunogen 13 amino acids near the carboxy terminus of human IL-36G. Nature Synthetic Expression System N/A Species Reactivity Human Conjugate Unconjugated Applications Used as a blocking peptide in immunoblotting applications. Procedure None Format Liquid Concentration 200 μg/mL Size 0.05mg Preservative None Storage -20°C ANTIGEN GENE INFORMATION Gene Name IL36G interleukin 36, gamma [ Homo sapiens (human) ] Official Symbol IL36G 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved Synonyms IL36G; interleukin -

Growth Factor Data Sheet
Growth Factor Data Sheet GoldBio growth factors are manufactured for RESEARCH USE ONLY and cannot be sold for human consumption! Interleukin 36B (IL36B) is a pro-inflammatory cytokine which plays an important role in the pathophysiology of several diseases. IL36A, IL36B, and IL36G (formerly IL1F6, IL1F8, and IL1F9) are all IL1 family members that signal through the IL1 receptor family members IL1Rrp2 (IL1RL2) and IL1RAcP. IL36B is reported to be expressed at higher levels in psoriatic plaques than in symptomless psoriatic skin or healthy control skin. IL36B can also stimulate the production of IL6 and IL8 in synovial fibroblasts, articular chondrocytes and mature adipocytes. Catalog Number 1310-36D Product Name IL36B (IL-36 beta), Murine (153 a.a.) Recombinant Murine Interleukin-36β IL36B, IL36β Interleukin 36β Interleukin 1 Family, Member 8 (IL1F8) Source Escherichia coli MW ~17.4 kDa (153 amino acids) Sequence RAASPSLRHV QDLSSRVWIL QNNILTAVPR KEQTVPVTIT LLPCQYLDTL ETNRGDPTYM GVQRPMSCLF CTKDGEQPVL QLGEGNIMEM YNKKEPVKAS LFYHKKSGTT STFESAAFPG WFIAVCSKGS CPLILTQELG EIFITDFEMI VVH Purity >95% by SDS-PAGE and HPLC analyses Biological Activity Fully biologically active when compared to standard. The ED50 as determined by inducing IL-6 secretion in murine NIH/3T3 cells is less than 25 ng/ml, corresponding to a specific activity of >4.0 × 104 IU/mg. Formulation Sterile filtered white lyophilized powder. Purified and tested for use in cell culture. Storage/Handling This lyophilized preparation is stable at 2-8°C, but should be kept at -20°C for long term storage. The reconstituted sample can be apportioned into working aliquots and stored at -80 °C for up to 6 months. -

The Role of Interleukin-1 Cytokine Family (IL-1Β, IL-37) and Interleukin-12 Cytokine
bioRxiv preprint doi: https://doi.org/10.1101/502609; this version posted December 22, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Title: 2 The Role of Interleukin-1 cytokine family (IL-1β, IL-37) and interleukin-12 cytokine 3 family (IL-12, IL-35) in eumycetoma infection pathogenesis. 4 5 6 Authors 7 Amir Abushouk1,2, Amre Nasr1,2,3, Emad Masuadi4, Gamal Allam5,6, Emmanuel E. Siddig7, 8 Ahmed H. Fahal7 9 10 1Department of Basic Medical Sciences, College of Medicine, King Saud Bin Abdul-Aziz 11 University for Health Sciences, Jeddah, Kingdom of Saudi Arabia. E. mail: shouka@ksau- 12 hs.edu.sa 13 14 2King Abdullah International Medical Research Centre, National Guard Health Affairs, 15 Kingdom of Saudi Arabia. 16 17 3Department of Microbiology, College of Sciences and Technology, Al-Neelain University, 18 P.O. Box 1027, Khartoum, Sudan. [email protected] 19 20 4Research Unit, Department of Medical Education, College of Medicine-Riyadh, King Saud 21 Bin Abdul-Aziz University for Health Sciences, Riyadh, Kingdom of Saudi Arabia. E. mail: 22 [email protected] 23 bioRxiv preprint doi: https://doi.org/10.1101/502609; this version posted December 22, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

IL-37 Is Increased in Brains of Children with Autism Spectrum Disorder and Inhibits Human Microglia Stimulated by Neurotensin
IL-37 is increased in brains of children with autism spectrum disorder and inhibits human microglia stimulated by neurotensin Irene Tsilionia, Arti B. Patela,b, Harry Pantazopoulosc,1, Sabina Berrettac, Pio Contid, Susan E. Leemane,2, and Theoharis C. Theoharidesa,b,f,2 aLaboratory of Molecular Immunopharmacology and Drug Discovery, Department of Immunology, Tufts University School of Medicine, Boston, MA 02111; bSackler School of Graduate Biomedical Sciences, Tufts University School of Medicine, Boston, MA 02111; cTranslational Neuroscience Laboratory, McLean Hospital, Harvard Medical School, Belmont, MA 02478; dImmunology Division, Postgraduate Medical School, University of Chieti, 65100 Pescara, Italy; eDepartment of Pharmacology, Boston University School of Medicine, Boston, MA 02118; and fDepartment of Internal Medicine, Tufts Medical Center, Tufts University School of Medicine, Boston, MA 02111 Contributed by Susan E. Leeman, July 15, 2019 (sent for review May 3, 2019; reviewed by Charles A. Dinarello and Richard E. Frye) Autism spectrum disorder (ASD) does not have a distinct pathogenesis to Toll-like receptor (TLR) activation. An IL-37 precursor (pro– or effective treatment. Increasing evidence supports the presence of IL-37) is cleaved by caspase-1 into mature IL-37, some of which immune dysfunction and inflammation in the brains of children with (∼20%) enters the nucleus and the rest is released along with the ASD. In this report, we present data that gene expression of the pro–IL-37 outside the cells (34) where both are biologically active. antiinflammatory cytokine IL-37, as well as of the proinflammatory Extracellular proteases can then process pro–IL-37 into a much cytokines IL-18 and TNF, is increased in the amygdala and dorsolateral more biologically active form as shown for the recombinant IL-37b prefrontal cortex of children with ASD as compared to non-ASD with the N terminus Val46 (V46-218) (35). -

Evolutionary Divergence and Functions of the Human Interleukin (IL) Gene Family Chad Brocker,1 David Thompson,2 Akiko Matsumoto,1 Daniel W
UPDATE ON GENE COMPLETIONS AND ANNOTATIONS Evolutionary divergence and functions of the human interleukin (IL) gene family Chad Brocker,1 David Thompson,2 Akiko Matsumoto,1 Daniel W. Nebert3* and Vasilis Vasiliou1 1Molecular Toxicology and Environmental Health Sciences Program, Department of Pharmaceutical Sciences, University of Colorado Denver, Aurora, CO 80045, USA 2Department of Clinical Pharmacy, University of Colorado Denver, Aurora, CO 80045, USA 3Department of Environmental Health and Center for Environmental Genetics (CEG), University of Cincinnati Medical Center, Cincinnati, OH 45267–0056, USA *Correspondence to: Tel: þ1 513 821 4664; Fax: þ1 513 558 0925; E-mail: [email protected]; [email protected] Date received (in revised form): 22nd September 2010 Abstract Cytokines play a very important role in nearly all aspects of inflammation and immunity. The term ‘interleukin’ (IL) has been used to describe a group of cytokines with complex immunomodulatory functions — including cell proliferation, maturation, migration and adhesion. These cytokines also play an important role in immune cell differentiation and activation. Determining the exact function of a particular cytokine is complicated by the influence of the producing cell type, the responding cell type and the phase of the immune response. ILs can also have pro- and anti-inflammatory effects, further complicating their characterisation. These molecules are under constant pressure to evolve due to continual competition between the host’s immune system and infecting organisms; as such, ILs have undergone significant evolution. This has resulted in little amino acid conservation between orthologous proteins, which further complicates the gene family organisation. Within the literature there are a number of overlapping nomenclature and classification systems derived from biological function, receptor-binding properties and originating cell type. -
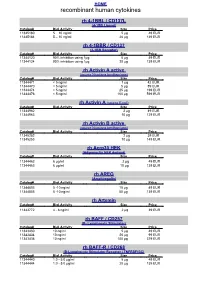
Recombinant Human Cytokines
HOME recombinant human cytokines rh 4-1BBL / CD137L (4-1BB Ligand) Catalog# Biol.Activity Size Price 11345180 5 – 10 ng/ml 5 µg 49 EUR 11345184 5 – 10 ng/ml 20 µg 139 EUR rh 4-1BBR / CD137 (4-1BB Receptor) Catalog# Biol.Activity Size Price 11344120 90% inhibition using 1µg 5 µg 49 EUR 11344124 90% inhibition using 1µg 20 µg 139 EUR rh Activin A active (source Nicotiana benthamiana) Catalog# Biol.Activity Size Price 11344471 < 5 ng/ml 1 µg 42 EUR 11344470 < 5 ng/ml 5 µg 89 EUR 11344474 < 5 ng/ml 25 µg 199 EUR 11344476 < 5 ng/ml 100 µg 599 EUR rh Activin A (source E.coli) Catalog# Biol.Activity Size Price 11344962 2 µg 49 EUR 11344963 10 µg 129 EUR rh Activin B active (source Nicotiana benthamiana) Catalog# Biol.Activity Size Price 11345252 2 µg 39 EUR 11345253 10 µg 149 EUR rh Acrp30 HEK (Adiponectin HEK derived) Catalog# Biol.Activity Size Price 11344462 6 µg/ml 2 µg 49 EUR 11344463 6 µg/ml 10 µg 139 EUR rh AREG (Amphiregulin) Catalog# Biol.Activity Size Price 11344803 5 -10 ng/ml 10 µg 49 EUR 11344805 5 -10 ng/ml 50 µg 139 EUR rh Artemin Catalog# Biol.Activity Size Price 11343772 4 - 8 ng/ml 2 µg 39 EUR rh BAFF / CD257 (B- Lymphocyte Stimulator) Catalog# Biol.Activity Size Price 11343430 10 ng/ml 5 µg 49 EUR 11343434 10 ng/ml 20 µg 99 EUR 11343436 10 ng/ml 100 µg 379 EUR rh BAFF-R / CD268 (B-Lymphocyte Stimulator Receptor / TNFRSF13C) Catalog# Biol.Activity Size Price 11344440 1.0 - 5.0 µg/ml 5 µg 49 EUR 11344444 1.0 - 5.0 µg/ml 20 µg 139 EUR HOME recombinant human cytokines rh BCA-1 (CXCL13) Catalog# Biol.Activity Size Price 11344180 -

Reduced Concentrations of the B Cell Cytokine Interleukin 38 Are Associated with Cardiovascular Disease Risk in Overweight Subjects
Eur. J. Immunol. 2020. 00: 1–10 DOI: 10.1002/eji.201948390 Dennis M. de Graaf et al. 1 Clinical Allergy and inflammation Research Article Reduced concentrations of the B cell cytokine interleukin 38 are associated with cardiovascular disease risk in overweight subjects DennisM.deGraaf1,2 , Martin Jaeger2 ,IngeC.L.vanden Munckhof2 , Rob ter Horst2 ,KikiSchraa2 , Jelle Zwaag3 , Matthijs Kox3 , Mayumi Fujita4 , Takeshi Yamauchi4, Laura Mercurio5 , Stefania Madonna5 , Joost H.W. Rutten2 , Jacqueline de Graaf2 ,NielsP.Riksen2 , Frank L. van de Veerdonk2 , Mihai G. Netea2 , Leo A.B. Joosten2 and Charles A. Dinarello1,2 1 Department of Medicine, University of Colorado Denver, Aurora, CO, USA 2 Department of Internal Medicine and Radboud Institute of Molecular Life Science (RIMLS), Radboud University Medical Center, Nijmegen, The Netherlands 3 Department of Intensive Care Medicine and Radboud Institute of Molecular Life Science (RIMLS), Radboud University Medical Center, Nijmegen, The Netherlands 4 Department of Dermatology, University of Colorado Denver, Aurora, CO, USA 5 Laboratory of Experimental Immunology, IDI-IRCCSFondazione Luigi M. Monti, Rome, Italy The IL-1 family member IL-38 (IL1F10) suppresses inflammatory and autoimmune con- ditions. Here, we report that plasma concentrations of IL-38 in 288 healthy Europeans correlate positively with circulating memory B cells and plasmablasts. IL-38 correlated negatively with age (p = 0.02) and was stable in 48 subjects for 1 year. In comparison with primary keratinocytes, IL1F10 expression in CD19+ B cells from PBMC was lower, whereas cell-associated IL-38 expression was comparable. In vitro, IL-38 is released from CD19+ B cells after stimulation with rituximab.