The Generation and Maintenance of Metabolic Alkalosis
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Pathophysiology of Acid Base Balance: the Theory Practice Relationship
Intensive and Critical Care Nursing (2008) 24, 28—40 ORIGINAL ARTICLE Pathophysiology of acid base balance: The theory practice relationship Sharon L. Edwards ∗ Buckinghamshire Chilterns University College, Chalfont Campus, Newland Park, Gorelands Lane, Chalfont St. Giles, Buckinghamshire HP8 4AD, United Kingdom Accepted 13 May 2007 KEYWORDS Summary There are many disorders/diseases that lead to changes in acid base Acid base balance; balance. These conditions are not rare or uncommon in clinical practice, but every- Arterial blood gases; day occurrences on the ward or in critical care. Conditions such as asthma, chronic Acidosis; obstructive pulmonary disease (bronchitis or emphasaemia), diabetic ketoacidosis, Alkalosis renal disease or failure, any type of shock (sepsis, anaphylaxsis, neurogenic, cardio- genic, hypovolaemia), stress or anxiety which can lead to hyperventilation, and some drugs (sedatives, opoids) leading to reduced ventilation. In addition, some symptoms of disease can cause vomiting and diarrhoea, which effects acid base balance. It is imperative that critical care nurses are aware of changes that occur in relation to altered physiology, leading to an understanding of the changes in patients’ condition that are observed, and why the administration of some immediate therapies such as oxygen is imperative. © 2007 Elsevier Ltd. All rights reserved. Introduction the essential concepts of acid base physiology is necessary so that quick and correct diagnosis can The implications for practice with regards to be determined and appropriate treatment imple- acid base physiology are separated into respi- mented. ratory acidosis and alkalosis, metabolic acidosis The homeostatic imbalances of acid base are and alkalosis, observed in patients with differing examined as the body attempts to maintain pH bal- aetiologies. -

Evaluation and Treatment of Alkalosis in Children
Review Article 51 Evaluation and Treatment of Alkalosis in Children Matjaž Kopač1 1 Division of Pediatrics, Department of Nephrology, University Address for correspondence Matjaž Kopač, MD, DSc, Division of Medical Centre Ljubljana, Ljubljana, Slovenia Pediatrics, Department of Nephrology, University Medical Centre Ljubljana, Bohoričeva 20, 1000 Ljubljana, Slovenia J Pediatr Intensive Care 2019;8:51–56. (e-mail: [email protected]). Abstract Alkalosisisadisorderofacid–base balance defined by elevated pH of the arterial blood. Metabolic alkalosis is characterized by primary elevation of the serum bicarbonate. Due to several mechanisms, it is often associated with hypochloremia and hypokalemia and can only persist in the presence of factors causing and maintaining alkalosis. Keywords Respiratory alkalosis is a consequence of dysfunction of respiratory system’s control ► alkalosis center. There are no pathognomonic symptoms. History is important in the evaluation ► children of alkalosis and usually reveals the cause. It is important to evaluate volemia during ► chloride physical examination. Treatment must be causal and prognosis depends on a cause. Introduction hydrogen ion concentration and an alkalosis is a pathologic Alkalosis is a disorder of acid–base balance defined by process that causes a decrease in the hydrogen ion concentra- elevated pH of the arterial blood. According to the origin, it tion. Therefore, acidemia and alkalemia indicate the pH can be metabolic or respiratory. Metabolic alkalosis is char- abnormality while acidosis and alkalosis indicate the patho- acterized by primary elevation of the serum bicarbonate that logic process that is taking place.3 can result from several mechanisms. It is the most common Regulation of hydrogen ion balance is basically similar to form of acid–base balance disorders. -

Oral Rehydration of Adult Cattle Using Isotonic Solution of Sugar, Sodium Chloride and Potassium Chloride
Haryana Vet. (Dec., 2019) 58(2), 166-169 Research Article ABSTRACT Fig 2: Transmission electron photomicrograph of monocyte of dog Present study comprised of 72 crossbred cows (group I= 60 endometritic and group II=12 healthy) at 30±2days postpartum. The showing heterochromatin (a), euchromatin (b), cytoplasmic process (c), polymorphonuclear neutrophils (PMN) cell coun Vacuole and nuclear membrane. Uranyl acetate and lead citrate × 25500 Figure 1: Cyclic conditions for PCR profiling for detection of Salmonella genes ASSOCIATION OF SEMEN TRAITS IN CONSECUTIVE EJACULATES WITH FSH-β GENE POLYMORPHISM IN HOLSTEIN-FRIESIAN CROSSBRED BULLS FROM INDIA VIJAY KADAM, ABH trus synchronizathod that synchronizes ovulations is Corresponding author: [email protected] Fig. 1. Histogram depicting frequency distribution of animal named briefly as “Ovsynch” (Pursley et al., 1995). The right score of respondents Clinical Article study was aimed to evaluate the efficacy of different methods of estrus sync Fig. 1. Semilogarithmic plot of plasma concentration time profile of amoxicillin and cloxacillin following single dose (10 mg/kg) i.v. and i.m. administration in sheep (n=4) Haryana Vet. (Dec., 2019) 58(2), 166-169 Research Article 2003) which might lead to increased chances of urolith the time for the urinary tract to restore patency (Parrah, Haryana Vet. (March, 2020) 59(SI), 93-95 Short Communication Research Article formation. The increased hospital incidence can also be 2009) in conjugation with supportive treatments like COMPARATIVE EFFICACY OF SYNCHRONIZATION PROTOCOLS FOR IMPROVING attributed to the proximity of the clinic as well. According to peritoneal lavage, urinary acidifiers and urinary ORAL REHYDRATION OF ADULT CATTLE USING ISOTONIC SOLUTION OF SUGAR, FERTILITY IN POSTPARTUM CROSSBRED DAIRY COWS data published by Department ff Soil Science, Haryana, antiseptics. -

TITLE: Acid-Base Disorders PRESENTER: Brenda Suh-Lailam
TITLE: Acid-Base Disorders PRESENTER: Brenda Suh-Lailam Slide 1: Hello, my name is Brenda Suh-Lailam. I am an Assistant Director of Clinical Chemistry and Mass Spectrometry at Ann & Robert H. Lurie Children’s Hospital of Chicago, and an Assistant Professor of Pathology at Northwestern Feinberg School of Medicine. Welcome to this Pearl of Laboratory Medicine on “Acid-Base Disorders.” Slide 2: During metabolism, the body produces hydrogen ions which affect metabolic processes if concentration is not regulated. To maintain pH within physiologic limits, there are several buffer systems that help regulate hydrogen ion concentration. For example, bicarbonate, plasma proteins, and hemoglobin buffer systems. The bicarbonate buffer system is the major buffer system in the blood. Slide 3: In the bicarbonate buffer system, bicarbonate, which is the metabolic component, is controlled by the kidneys. Carbon dioxide is the respiratory component and is controlled by the lungs. Changes in the respiratory and metabolic components, as depicted here, can lead to a decrease in pH termed acidosis, or an increase in pH termed alkalosis. Slide 4: Because the bicarbonate buffer system is the major buffer system of blood, estimation of pH using the Henderson-Hasselbalch equation is usually performed, expressed as a ratio of bicarbonate and carbon dioxide. Where pKa is the pH at which the concentration of protonated and unprotonated species are equal, and 0.0307 is the solubility coefficient of carbon dioxide. Four variables are present in this equation; knowing three variables allows for calculation of the fourth. Since pKa is a constant, and pH and carbon dioxide are measured during blood gas analysis, bicarbonate can, therefore, be determined using this equation. -

Package Insert Template for Oral Rehydration Salt (Ors)
PACKAGE INSERT TEMPLATE FOR ORAL REHYDRATION SALT (ORS) Brand or Product Name [Product name] Powder for Oral Solution [Product name] Liquid in the form of solution/suspension Name and Strength of Active Substance(s) Sodium chloride ………………(12.683% w/v) Glucose, anhydrous…………...(65.854% w/v) Potassium chloride………...…...(7.317% w/v) Trisodium citrate, dihydrate ….(14.146% w/v) Product Description [Visual description of the appearance of the product (eg colour etc)] Eg:A white to off-white colour granules, when dissolved in water, forms an orange colour solution. Pharmacodynamics The reconstituted solution contains a mixture of sodium and potassium salts along with glucose, which facilitates the absorption of sodium and potassium from the intestine. Water is drawn from the bowel by the osmotic effect. As well as “drying up” the stools, the dehydration and loss of electrolytes caused by the diarrhoea is corrected by the water and electrolytes absorbed. Pharmacokinetics Glucose After oral administration glucose is completely absorbed by a sodium dependent uptake mechanism exhibiting saturation kinetics. Blood levels return to normal within two hours of ingestion. Potassium Chloride No specific control mechanisms limit absorption of potassium, which is usually complete. Potassium is excreted largely by the kidneys, though 10% is excreted by the colonic mucosa. Potassium excretion is reduced in patients with renal impairment and in the elderly, so extreme caution should be used in treating such patients with potassium salts. Sodium Bicarbonate Kinetics are determined by the physiological state of the patient at the time. Sodium Chloride Readily absorbed from the gastrointestinal tract. Gut absorption, particularly in the jejunum is enhanced by the addition of glucose. -

Diabetic Ketoalkalosis in Children and Adults
Original Article Diabetic Ketoalkalosis in Children and Adults Emily A. Huggins, MD, Shawn A. Chillag, MD, Ali A. Rizvi, MD, Robert R. Moran, PhD, and Martin W. Durkin, MD, MPH and DR are calculated because the pH and bicarbonate may be near Objectives: Diabetic ketoacidosis (DKA) with metabolic alkalosis normal or even elevated. In addition to having interesting biochemical (diabetic ketoalkalosis [DKALK]) in adults has been described in the features as a complex acid-base disorder, DKALK can pose diagnostic literature, but not in the pediatric population. The discordance in the and/or therapeutic challenges. change in the anion gap (AG) and the bicarbonate is depicted by an Key Words: delta ratio, diabetic ketoacidosis, diabetic ketoalkalosis, elevated delta ratio (DR; rise in AG/drop in bicarbonate), which is metabolic alkalosis normally approximately 1. The primary aim of this study was to de- termine whether DKALK occurs in the pediatric population, as has been seen previously in the adult population. The secondary aim was iabetic ketoacidosis (DKA), a common and serious dis- to determine the factors that may be associated with DKALK. Dorder that almost always results in hospitalization, is de- Methods: A retrospective analysis of adult and pediatric cases with a fined by the presence of hyperglycemia, reduced pH, metabolic 1 primary or secondary discharge diagnosis of DKA between May 2008 and acidosis, elevated anion gap (AG), and serum or urine ketones. August 2010 at a large urban hospital was performed. DKALK was as- In some situations, a metabolic alkalosis coexists with DKA sumedtobepresentiftheDRwas91.2 or in cases of elevated bicarbonate. -

Parenteral Nutrition Primer: Balance Acid-Base, Fluid and Electrolytes
Parenteral Nutrition Primer: Balancing Acid-Base, Fluids and Electrolytes Phil Ayers, PharmD, BCNSP, FASHP Todd W. Canada, PharmD, BCNSP, FASHP, FTSHP Michael Kraft, PharmD, BCNSP Gordon S. Sacks, Pharm.D., BCNSP, FCCP Disclosure . The program chair and presenters for this continuing education activity have reported no relevant financial relationships, except: . Phil Ayers - ASPEN: Board Member/Advisory Panel; B Braun: Consultant; Baxter: Consultant; Fresenius Kabi: Consultant; Janssen: Consultant; Mallinckrodt: Consultant . Todd Canada - Fresenius Kabi: Board Member/Advisory Panel, Consultant, Speaker's Bureau • Michael Kraft - Rockwell Medical: Consultant; Fresenius Kabi: Advisory Board; B. Braun: Advisory Board; Takeda Pharmaceuticals: Speaker’s Bureau (spouse) . Gordon Sacks - Grant Support: Fresenius Kabi Sodium Disorders and Fluid Balance Gordon S. Sacks, Pharm.D., BCNSP Professor and Department Head Department of Pharmacy Practice Harrison School of Pharmacy Auburn University Learning Objectives Upon completion of this session, the learner will be able to: 1. Differentiate between hypovolemic, euvolemic, and hypervolemic hyponatremia 2. Recommend appropriate changes in nutrition support formulations when hyponatremia occurs 3. Identify drug-induced causes of hypo- and hypernatremia No sodium for you! Presentation Outline . Overview of sodium and water . Dehydration vs. Volume Depletion . Water requirements & Equations . Hyponatremia • Hypotonic o Hypovolemic o Euvolemic o Hypervolemic . Hypernatremia • Hypovolemic • Euvolemic • Hypervolemic Sodium and Fluid Balance . Helpful hint: total body sodium determines volume status, not sodium status . Examples of this concept • Hypervolemic – too much volume • Hypovolemic – too little volume • Euvolemic – normal volume Water Distribution . Total body water content varies from 50-70% of body weight • Dependent on lean body mass: fat ratio o Fat water content is ~10% compared to ~75% for muscle mass . -

Observations Compensatory Hypochloraemic Alkalosis In
Letters 871 tended to the DQ-LTR13 integration, where we also find a con- 2. Pascual M, Martin J, Nieto A et al. (2001) Distribution of tribution of DQ-LTR13 to RA susceptibility in DQ8-positive HERV-LTR elements in the 5′-flanking region of HLA- individuals. Furthermore we recently published that patients DQB1 and association with autoimmunity. Immunogenetics with Addison’s disease—another HLA DQ8-associated autoim- 53:114–118 mune disease of the adrenals which may occur in combination 3. Donner H, Tonjes RR, Bontrop RE et al. (1999) Intronic se- with Type 1 diabetes as part of the autoimmune pluriglandular quence motifs of HLA-DQB1 are shared between humans, syndrome Type 2—have more often the DQ8-DQ-LTR13 com- apes and Old World monkeys, but a retroviral LTR element bination in contrast to the DQ-LTR3 insertion. Although both (DQLTR3) is human specific. Tissue Antigens 53:551–558 DQ-LTR3 and DQ-LTR13 are linked to DQ8, DQ-LTR13 en- 4. Donner H, Tonjes RR, Van der Auwera B et al. (1999) The hances the risk for Addison’s disease [7]. presence or absence of a retroviral long terminal repeat Whether DQ-LTR13 has a functional significance or not is influences the genetic risk for type 1 diabetes conferred by currently under investigation. Preliminary data indicate that human leukocyte antigen DQ haplotypes. J Clin Endocrinol DQ-LTR13 harbours regulatory capacity and shows functional Metab 84:1404–1408 activity of an upstream element as revealed by analyses of nu- 5. Seidl C, Donner H, Petershofen E et al. -

A Case-Based Approach to Acid-Base Disorders
A Case-Based Approach to Acid-Base Disorders Justin Muir, PharmD Clinical Pharmacy Manager, Medical ICU NewYork-Presbyterian Hospital Columbia University Irving Medical Center [email protected] Disclosures None Objectives At the completion of this activity, pharmacists will be able to: 1. Describe acid-base physiology and disease states that lead to acid-base disorders. 2. Demonstrate a step-wise approach to interpretation of acid-base disorders and compensatory states. 3. Analyze contemporary literature regarding the use of sodium bicarbonate in metabolic acidosis. At the completion of this activity, pharmacy technicians will be able to: 1. Explain the importance of acid-base balance. 2. List the acid-base disorders seen in clinical practice. 3. Identify potential therapies used to treat acid-base disorders. Case A 51 year old man with history of erosive esophagitis, diabetes mellitus, chronic pancreatitis, and bipolar disorder is admitted with several days of severe nausea, vomiting, and abdominal pain. 135 87 31 pH 7.46 / pCO 29 / pO 81 861 2 2 BE -3.8 / HCO - 18 / SaO 96 5.6 20 0.9 3 2 • What additional data should be obtained? • What acid base disturbance(s) is/are present? Introduction • Acid base status is tightly regulated to maintain normal biochemical reactions and organ function • Body uses multiple mechanisms to maintain homeostasis • Abnormalities are extremely common in hospitalized patients with a higher incidence in critically ill with more complex pictures • A standard approach to analysis can help guide diagnosis and treatment Important acid-base determinants Blood gas generally includes at least: Normal range Measurement Description (arterial blood) pH -log [H+] 7.35-7.45 pCO2 partial pressure of dissolved CO2 35-45 mmHg pO2 partial pressure of dissolved O2 80-100 mmHg Base excess calculated measure of metabolic acid/base deviation from normal -3 to +3 SO2 calculated measure of Hgb O2 saturation based on pO2 95-100% - HCO3 calculated measure based on relationship of pH and pCO2 22-26 mEq/L Haber RJ. -

ACS/ASE Medical Student Core Curriculum Acid-Base Balance
ACS/ASE Medical Student Core Curriculum Acid-Base Balance ACID-BASE BALANCE Epidemiology/Pathophysiology Understanding the physiology of acid-base homeostasis is important to the surgeon. The two acid-base buffer systems in the human body are the metabolic system (kidneys) and the respiratory system (lungs). The simultaneous equilibrium reactions that take place to maintain normal acid-base balance are: H" HCO* ↔ H CO ↔ H O l CO g To classify the type of disturbance, a blood gas (preferably arterial) and basic metabolic panel must be obtained. A basic understanding of normal acid-base buffer physiology is required to understand alterations in these labs. The normal pH of human blood is 7.40 (7.35-7.45). This number is tightly regulated by the two buffer systems mentioned above. The lungs contain carbonic anhydrase which is capable of converting carbonic acid to water and CO2. The respiratory response results in an alteration to ventilation which allows acid to be retained or expelled as CO2. Therefore, bradypnea will result in respiratory acidosis while tachypnea will result in respiratory alkalosis. The respiratory buffer system is fast acting, resulting in respiratory compensation within 30 minutes and taking approximately 12 to 24 hours to reach equilibrium. The renal metabolic response results in alterations in bicarbonate excretion. This system is more time consuming and can typically takes at least three to five days to reach equilibrium. Five primary classifications of acid-base imbalance: • Metabolic acidosis • Metabolic alkalosis • Respiratory acidosis • Respiratory alkalosis • Mixed acid-base disturbance It is important to remember that more than one of the above processes can be present in a patient at any given time. -

Practical Fluid and Electrolyte Therapy and Its Pathophysiological Basis
There are few field tests with proven reliability for Laboratory procedures that may serve adequately on-the-farm analysis. This is not to imply that field for clinical diagnosis may not necessarily be adequate tests are of no value but reflects a lack of information for evidence in a lawsuit. This should be recognized and data concerning their usefulness. The prior to rendering any opinion to the client concer diphenylamine test for nitrates will detect high con ning a basis for suing for damages or recovery of centration of nitrate (2%) in feed but is not sufficient losses. ly sensitive for nitrate concentrations (0.5%) that The future direction of clinical toxicology is depen dent on greater specialization of veterinary tox may lead to chronic nitrate intoxication. The reagent icologists and subsidized support of university and is made by adding 0.5 gm of diphenylamine to 20 ml governmental analytical laboratories to serve in a water and concentrated sulfuric acid is added q.s. to consultative capacity with the private veterinary 100 ml. This is a stock solution which is mixed with practitioner. It is impossible to speculate on the im equal parts of 80% sulfuric acid for use. One drop of pact of chronic intoxication on animal health because the reagent is placed on the cut surface of the plant of our limited ability for diagnosis. Since we institued and a color change from green to blue is indicative of a modest program of lead analysis in our laboratory high nitrate content (approximately 2%). Corn there has been a significant increase in the number of suspected of containing aflatoxin may be examined diagnoses of chronic lead intoxication in both pet with an ultraviolet light. -
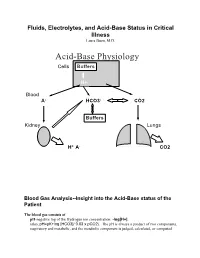
Acid-Base Physiology Cells Buffers
Fluids, Electrolytes, and Acid-Base Status in Critical Illness Laura Ibsen, M.D. Acid-Base Physiology Cells Buffers H+ Blood A- HCO3- CO2 Buffers Kidney Lungs H+ A- CO2 Blood Gas Analysis--Insight into the Acid-Base status of the Patient The blood gas consists of pH-negative log of the Hydrogen ion concentration: -log[H+]. (also, pH=pK+log [HCO3]/ 0.03 x pCO2). The pH is always a product of two components, respiratory and metabolic, and the metabolic component is judged, calculated, or computed by allowing for the effect of the pCO2, ie, any change in the pH unexplained by the pCO2 indicates a metabolic abnormality. - + CO2+H20º H2CO3ºHCO3 + H CO2 and water form carbonic acid or H2CO3, which is in equilibrium with bicarbonate (HCO3-)and hydrogen ions (H+). A change in the concentration of the reactants on either side of the equation affects the subsequent direction of the reaction. For example, an increase in CO2 will result in increased carbonic acid formation (H2CO3) which leads to an increase in both HCO3- and H+ (\pH). Normally, at pH 7.4, a ratio of one part carbonic acid to twenty parts bicarbonate is present in the extracellular fluid [HCO3-/H2CO3]=20. A change in the ratio will affect the pH of the fluid. If both components change (ie, with chronic compensation), the pH may be normal, but the other components will not. pCO2-partial pressure of carbon dioxide. Hypoventilation or hyperventilation (ie, minute ventilation--tidal volume x respitatory rate--imperfectly matched to physiologic demands) will lead to elevation or depression, respectively, in the pCO2.