Standards in Genomic Sciences 2014, 9:1
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Food Waste Composting and Microbial Community Structure Profiling
processes Review Food Waste Composting and Microbial Community Structure Profiling Kishneth Palaniveloo 1,* , Muhammad Azri Amran 1, Nur Azeyanti Norhashim 1 , Nuradilla Mohamad-Fauzi 1,2, Fang Peng-Hui 3, Low Hui-Wen 3, Yap Kai-Lin 3, Looi Jiale 3, Melissa Goh Chian-Yee 3, Lai Jing-Yi 3, Baskaran Gunasekaran 3,* and Shariza Abdul Razak 4,* 1 Institute of Ocean and Earth Sciences, Institute for Advanced Studies Building, University of Malaya, Wilayah Persekutuan Kuala Lumpur 50603, Malaysia; [email protected] (M.A.A.); [email protected] (N.A.N.) 2 Institute of Biological Sciences, Faculty of Science, University of Malaya, Wilayah Persekutuan Kuala Lumpur 50603, Malaysia; [email protected] 3 Faculty of Applied Science, UCSI University (South Wing), Cheras, Wilayah Persekutuan Kuala Lumpur 56000, Malaysia; [email protected] (F.P.-H.); [email protected] (L.H.-W.); [email protected] (Y.K.-L.); [email protected] (L.J.); [email protected] (M.G.C.-Y.); [email protected] (L.J.-Y.) 4 Nutrition and Dietetics Program, School of Health Sciences, Health Campus, Universiti Sains Malaysia, Kubang Kerian 16150, Kelantan, Malaysia * Correspondence: [email protected] (K.P.); [email protected] (B.G.); [email protected] (S.A.R.); Tel.: +60-3-7967-4640 (K.P.); +60-16-323-4159 (B.G.); +60-19-964-4043 (S.A.R.) Received: 20 May 2020; Accepted: 16 June 2020; Published: 22 June 2020 Abstract: Over the last decade, food waste has been one of the major issues globally as it brings a negative impact on the environment and health. -

Phylogeny of Nitrogenase Structural and Assembly Components Reveals New Insights Into the Origin and Distribution of Nitrogen Fixation Across Bacteria and Archaea
microorganisms Article Phylogeny of Nitrogenase Structural and Assembly Components Reveals New Insights into the Origin and Distribution of Nitrogen Fixation across Bacteria and Archaea Amrit Koirala 1 and Volker S. Brözel 1,2,* 1 Department of Biology and Microbiology, South Dakota State University, Brookings, SD 57006, USA; [email protected] 2 Department of Biochemistry, Genetics and Microbiology, University of Pretoria, Pretoria 0004, South Africa * Correspondence: [email protected]; Tel.: +1-605-688-6144 Abstract: The phylogeny of nitrogenase has only been analyzed using the structural proteins NifHDK. As nifHDKENB has been established as the minimum number of genes necessary for in silico predic- tion of diazotrophy, we present an updated phylogeny of diazotrophs using both structural (NifHDK) and cofactor assembly proteins (NifENB). Annotated Nif sequences were obtained from InterPro from 963 culture-derived genomes. Nif sequences were aligned individually and concatenated to form one NifHDKENB sequence. Phylogenies obtained using PhyML, FastTree, RapidNJ, and ASTRAL from individuals and concatenated protein sequences were compared and analyzed. All six genes were found across the Actinobacteria, Aquificae, Bacteroidetes, Chlorobi, Chloroflexi, Cyanobacteria, Deferribacteres, Firmicutes, Fusobacteria, Nitrospira, Proteobacteria, PVC group, and Spirochaetes, as well as the Euryarchaeota. The phylogenies of individual Nif proteins were very similar to the overall NifHDKENB phylogeny, indicating the assembly proteins have evolved together. Our higher resolution database upheld the three cluster phylogeny, but revealed undocu- Citation: Koirala, A.; Brözel, V.S. mented horizontal gene transfers across phyla. Only 48% of the 325 genera containing all six nif genes Phylogeny of Nitrogenase Structural and Assembly Components Reveals are currently supported by biochemical evidence of diazotrophy. -

From Genotype to Phenotype: Inferring Relationships Between Microbial Traits and Genomic Components
From genotype to phenotype: inferring relationships between microbial traits and genomic components Inaugural-Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakult¨at der Heinrich-Heine-Universit¨atD¨usseldorf vorgelegt von Aaron Weimann aus Oberhausen D¨usseldorf,29.08.16 aus dem Institut f¨urInformatik der Heinrich-Heine-Universit¨atD¨usseldorf Gedruckt mit der Genehmigung der Mathemathisch-Naturwissenschaftlichen Fakult¨atder Heinrich-Heine-Universit¨atD¨usseldorf Referent: Prof. Dr. Alice C. McHardy Koreferent: Prof. Dr. Martin J. Lercher Tag der m¨undlichen Pr¨ufung: 24.02.17 Selbststandigkeitserkl¨ arung¨ Hiermit erkl¨areich, dass ich die vorliegende Dissertation eigenst¨andigund ohne fremde Hilfe angefertig habe. Arbeiten Dritter wurden entsprechend zitiert. Diese Dissertation wurde bisher in dieser oder ¨ahnlicher Form noch bei keiner anderen Institution eingereicht. Ich habe bisher keine erfolglosen Promotionsversuche un- ternommen. D¨usseldorf,den . ... ... ... (Aaron Weimann) Statement of authorship I hereby certify that this dissertation is the result of my own work. No other person's work has been used without due acknowledgement. This dissertation has not been submitted in the same or similar form to other institutions. I have not previously failed a doctoral examination procedure. Summary Bacteria live in almost any imaginable environment, from the most extreme envi- ronments (e.g. in hydrothermal vents) to the bovine and human gastrointestinal tract. By adapting to such diverse environments, they have developed a large arsenal of enzymes involved in a wide variety of biochemical reactions. While some such enzymes support our digestion or can be used for the optimization of biotechnological processes, others may be harmful { e.g. mediating the roles of bacteria in human diseases. -

Functional Analysis of Multiple Nifb Genes of Paenibacillus Strains in Synthesis of Mo-, Fe- and V- Nitrogenase
Functional Analysis of Multiple nifB Genes of Paenibacillus Strains in Synthesis of Mo-, Fe- and V- Nitrogenase Qin Li China Agricultural University Haowei Zhang China Agricultural University Liqun Zhang China Agricultural University San-Feng Chen ( [email protected] ) China Agricultural University https://orcid.org/0000-0003-2956-9025 Research Keywords: Paenibacillus, nifB gene, Mo-nitrogenase, alternative nitrogenases Posted Date: April 22nd, 2021 DOI: https://doi.org/10.21203/rs.3.rs-444251/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published at Microbial Cell Factories on July 19th, 2021. See the published version at https://doi.org/10.1186/s12934-021-01629-9. Page 1/20 Abstract Background: Biological nitrogen xation is catalyzed by Mo-, V- and Fe-nitrogenases that are encoded by nif, vnf and anf genes, respectively. NifB is the key protein in synthesis of the cofactors of all nitrogenases. Most diazotrophic Paenibacillus strains have only one nifB gene located in a compact nif gene cluster (nifBHDKENX(orf1)hesAnifV). But some Paenibacillus strains have multiple nifB genes and their functions are not known. Results: We have analyzed the genomes of the 116 diazotrophic Paenibacillus strains and found that some Paenibacillus strains have 2-4 nifB genes. Phylogeny analysis shows that all nifB genes in Paenibacillus fall into 4 subclasses: the nifB1 being the rst gene within the compact nif gene cluster, the nifB2 being adjacent to anf or vnf genes, the other nifB3 and nifB4 being scattered on genomes. -

Identification and Classification of Known and Putative Antimicrobial Compounds Produced by a Wide Variety of Bacillales Species Xin Zhao1,2 and Oscar P
Zhao and Kuipers BMC Genomics (2016) 17:882 DOI 10.1186/s12864-016-3224-y RESEARCH ARTICLE Open Access Identification and classification of known and putative antimicrobial compounds produced by a wide variety of Bacillales species Xin Zhao1,2 and Oscar P. Kuipers1* Abstract Background: Gram-positive bacteria of the Bacillales are important producers of antimicrobial compounds that might be utilized for medical, food or agricultural applications. Thanks to the wide availability of whole genome sequence data and the development of specific genome mining tools, novel antimicrobial compounds, either ribosomally- or non-ribosomally produced, of various Bacillales species can be predicted and classified. Here, we provide a classification scheme of known and putative antimicrobial compounds in the specific context of Bacillales species. Results: We identify and describe known and putative bacteriocins, non-ribosomally synthesized peptides (NRPs), polyketides (PKs) and other antimicrobials from 328 whole-genome sequenced strains of 57 species of Bacillales by using web based genome-mining prediction tools. We provide a classification scheme for these bacteriocins, update the findings of NRPs and PKs and investigate their characteristics and suitability for biocontrol by describing per class their genetic organization and structure. Moreover, we highlight the potential of several known and novel antimicrobials from various species of Bacillales. Conclusions: Our extended classification of antimicrobial compounds demonstrates that Bacillales provide a rich source of novel antimicrobials that can now readily be tapped experimentally, since many new gene clusters are identified. Keywords: Antimicrobials, Bacillales, Bacillus, Genome-mining, Lanthipeptides, Sactipeptides, Thiopeptides, NRPs, PKs Background (bacteriocins) [4], as well as non-ribosomally synthesized Most of the species of the genus Bacillus and related peptides (NRPs) and polyketides (PKs) [5]. -

Genome Diversity of Spore-Forming Firmicutes MICHAEL Y
Genome Diversity of Spore-Forming Firmicutes MICHAEL Y. GALPERIN National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD 20894 ABSTRACT Formation of heat-resistant endospores is a specific Vibrio subtilis (and also Vibrio bacillus), Ferdinand Cohn property of the members of the phylum Firmicutes (low-G+C assigned it to the genus Bacillus and family Bacillaceae, Gram-positive bacteria). It is found in representatives of four specifically noting the existence of heat-sensitive vegeta- different classes of Firmicutes, Bacilli, Clostridia, Erysipelotrichia, tive cells and heat-resistant endospores (see reference 1). and Negativicutes, which all encode similar sets of core sporulation fi proteins. Each of these classes also includes non-spore-forming Soon after that, Robert Koch identi ed Bacillus anthracis organisms that sometimes belong to the same genus or even as the causative agent of anthrax in cattle and the species as their spore-forming relatives. This chapter reviews the endospores as a means of the propagation of this orga- diversity of the members of phylum Firmicutes, its current taxon- nism among its hosts. In subsequent studies, the ability to omy, and the status of genome-sequencing projects for various form endospores, the specific purple staining by crystal subgroups within the phylum. It also discusses the evolution of the violet-iodine (Gram-positive staining, reflecting the pres- Firmicutes from their apparently spore-forming common ancestor ence of a thick peptidoglycan layer and the absence of and the independent loss of sporulation genes in several different lineages (staphylococci, streptococci, listeria, lactobacilli, an outer membrane), and the relatively low (typically ruminococci) in the course of their adaptation to the saprophytic less than 50%) molar fraction of guanine and cytosine lifestyle in a nutrient-rich environment. -

Comparative Genomics and Phylogenomic Analyses of Lysine Riboswitch Distributions in Bacteria
RESEARCH ARTICLE Comparative genomics and phylogenomic analyses of lysine riboswitch distributions in bacteria Sumit Mukherjee1, Danny Barash2, Supratim Sengupta1* 1 Department of Physical Sciences, Indian Institute of Science Education and Research Kolkata, Mohanpur, India, 2 Department of Computer Science, Ben-Gurion University of the Negev, Beer-Sheva, Israel * [email protected] a1111111111 a1111111111 a1111111111 Abstract a1111111111 a1111111111 Riboswitches are cis-regulatory elements that regulate the expression of genes involved in biosynthesis or transport of a ligand that binds to them. Among the nearly 40 classes of riboswitches discovered so far, three are known to regulate the concentration of biologically encoded amino acids glycine, lysine, and glutamine. While some comparative genomics studies of riboswitches focusing on their gross distribution across different bacterial taxa OPEN ACCESS have been carried out recently, systematic functional annotation and analysis of lysine ribos- Citation: Mukherjee S, Barash D, Sengupta S (2017) Comparative genomics and phylogenomic witches and the genes they regulate are still lacking. We analyzed 2785 complete bacterial analyses of lysine riboswitch distributions in genome sequences to systematically identify 468 lysine riboswitches (not counting hits from bacteria. PLoS ONE 12(9): e0184314. https://doi. multiple strains of the same species) and obtain a detailed phylogenomic map of gene-spe- org/10.1371/journal.pone.0184314 cific lysine riboswitch distribution across diverse prokaryotic phyla. We find that lysine ribos- Editor: Young-Hwa Song, Fachhochschule Lubeck, witches are most abundant in Firmicutes and Gammaproteobacteria where they are found GERMANY upstream to both biosynthesis and/or transporter genes. They are relatively rare in all other Received: March 14, 2017 prokaryotic phyla where if present they are primarily found upstream to operons containing Accepted: August 22, 2017 many lysine biosynthesis genes. -

Phase II Final Report
FINAL REPORT Developing and Field-Testing Genetic Catabolic Probes for Monitored Natural Attenuation of 1,4-Dioxane SERDP Project ER-2301 SEPTEMBER 2019 Pedro Alvarez Ya He Mengyan Li Yu Yang Marcio Busi Da Silva Jacques Mathieu Rice University Distribution Statement A Page Intentionally Left Blank This report was prepared under contract to the Department of Defense Strategic Environmental Research and Development Program (SERDP). The publication of this report does not indicate endorsement by the Department of Defense, nor should the contents be construed as reflecting the official policy or position of the Department of Defense. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the Department of Defense. Page Intentionally Left Blank Form Approved REPORT DOCUMENTATION PAGE OMB No. 0704-0188 Public reporting burden for this collection of information is estimated to average 1 hour per response, including the time for reviewing instructions, searching existing data sources, gathering and maintaining the data needed, and completing and reviewing this collection of information. Send comments regarding this burden estimate or any other aspect of this collection of information, including suggestions for reducing this burden to Department of Defense, Washington Headquarters Services, Directorate for Information Operations and Reports (0704-0188), 1215 Jefferson Davis Highway, Suite 1204, Arlington, VA 22202- 4302. Respondents should be aware that notwithstanding any other provision of law, no person shall be subject to any penalty for failing to comply with a collection of information if it does not display a currently valid OMB control number. -
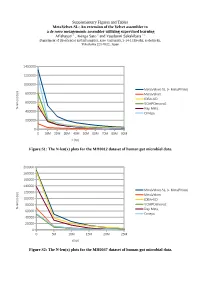
Supplementary Figures and Tables Metavelvet-SL
Supplementary Figures and Tables MetaVelvet-SL: An extension of the Velvet assembler to a de novo metagenomic assembler utilizing supervised learning Afiahayati 1 , Kengo Sato 1 and Yasubumi Sakakibara 1∗ Department of Biosciences and Informatics, Keio University, 3-14-1 Hiyoshi, Kohoku-ku, Yokohama 223-8522, Japan 1400000 1200000 1000000 MetaVelvet-SL (+ MetaPhlAn) ) p 800000 b MetaVelvet ( ) x ( IDBA-UD n 600000 e SOAPDenovo2 l - N Ray Meta 400000 Omega 200000 0 0 10M 20M 30M 40M 50M 60M 70M 80M 90M x (bp) Figure S1: The N-len(x) plots for the MH0012 dataset of human gut microbial data. 200000 180000 160000 140000 MetaVelvet-SL (+ MetaPhlAn) ) 120000 p b MetaVelvet ( ) 100000 x IDBA-UD ( n e l 80000 SOAPDenovo2 - N Ray Meta 60000 Omega 40000 20000 0 0 5M 10M 15M 20M 25M x(bp) Figure S2: The N-len(x) plots for the MH0047 dataset of human gut microbial data. 600000 500000 400000 MetaVelvet-SL (+ MetaPhlAn) ) p b MetaVelvet ( ) 300000 x IDBA-UD ( n e l SOAPDenovo2 - N 200000 Ray Meta Omega 100000 0 0 10M 20M 30M 40M 50M 60M 70M 80M 90M x (bp) Figure S3: The N-len(x) plots for the SRS017227 dataset of human gut microbial data. 800000 700000 600000 500000 MetaVelvet-SL (+ MetaPhlAn) ) p b MetaVelvet ( ) 400000 x IDBA-UD ( n e l SOAPDenovo2 - 300000 N Ray Meta 200000 Omega 100000 0 0 5M 10M 15M 20M 25M 30M 35M 40M 45M x (bp) Figure S4: The N-len(x) plots for the SRS018661 dataset of human gut microbial data. Formula to identify unique nodes in Velvet (Zerbino and Birney, 2008) ̄x2 ρ2− log2 2 F ( ̄x ,n,ρ )= +n 2 2 where ̄x = the coverage of node ρ = expected coverage of subgraph n = the length of node A node is “unique”, if its F > 5 . -

Reorganising the Order Bacillales Through Phylogenomics
Systematic and Applied Microbiology 42 (2019) 178–189 Contents lists available at ScienceDirect Systematic and Applied Microbiology jou rnal homepage: http://www.elsevier.com/locate/syapm Reorganising the order Bacillales through phylogenomics a,∗ b c Pieter De Maayer , Habibu Aliyu , Don A. Cowan a School of Molecular & Cell Biology, Faculty of Science, University of the Witwatersrand, South Africa b Technical Biology, Institute of Process Engineering in Life Sciences, Karlsruhe Institute of Technology, Germany c Centre for Microbial Ecology and Genomics, University of Pretoria, South Africa a r t i c l e i n f o a b s t r a c t Article history: Bacterial classification at higher taxonomic ranks such as the order and family levels is currently reliant Received 7 August 2018 on phylogenetic analysis of 16S rRNA and the presence of shared phenotypic characteristics. However, Received in revised form these may not be reflective of the true genotypic and phenotypic relationships of taxa. This is evident in 21 September 2018 the order Bacillales, members of which are defined as aerobic, spore-forming and rod-shaped bacteria. Accepted 18 October 2018 However, some taxa are anaerobic, asporogenic and coccoid. 16S rRNA gene phylogeny is also unable to elucidate the taxonomic positions of several families incertae sedis within this order. Whole genome- Keywords: based phylogenetic approaches may provide a more accurate means to resolve higher taxonomic levels. A Bacillales Lactobacillales suite of phylogenomic approaches were applied to re-evaluate the taxonomy of 80 representative taxa of Bacillaceae eight families (and six family incertae sedis taxa) within the order Bacillales. -

Microbial Life Cycles Link Global Modularity in Regulation to Mosaic Evolution
ARTICLES https://doi.org/10.1038/s41559-019-0939-6 Microbial life cycles link global modularity in regulation to mosaic evolution Jordi van Gestel 1,2,3,4*, Martin Ackermann3,4 and Andreas Wagner 1,2,5* Microbes are exposed to changing environments, to which they can respond by adopting various lifestyles such as swimming, colony formation or dormancy. These lifestyles are often studied in isolation, thereby giving a fragmented view of the life cycle as a whole. Here, we study lifestyles in the context of this whole. We first use machine learning to reconstruct the expression changes underlying life cycle progression in the bacterium Bacillus subtilis, based on hundreds of previously acquired expres- sion profiles. This yields a timeline that reveals the modular organization of the life cycle. By analysing over 380 Bacillales genomes, we then show that life cycle modularity gives rise to mosaic evolution in which life stages such as motility and sporu- lation are conserved and lost as discrete units. We postulate that this mosaic conservation pattern results from habitat changes that make these life stages obsolete or detrimental. Indeed, when evolving eight distinct Bacillales strains and species under laboratory conditions that favour colony growth, we observe rapid and parallel losses of the sporulation life stage across spe- cies, induced by mutations that affect the same global regulator. We conclude that a life cycle perspective is pivotal to under- standing the causes and consequences of modularity in both regulation and evolution. icrobes express an incredible range of lifestyles, from the We start our analysis by synthesizing data from previous stud- myriad of planktonic life forms floating in the oceans ies on the global transcription network of B. -
Improving the Cathodic Biofilm Growth Capabilities of Kyrpidia Spormannii
microorganisms Article Improving the Cathodic Biofilm Growth Capabilities of Kyrpidia spormannii EA-1 by Undirected Mutagenesis Tobias Jung 1, Max Hackbarth 2, Harald Horn 2 and Johannes Gescher 1,3,* 1 Department of Applied Biology, Institute for Applied Biosciences, Karlsruhe Institute of Technology (KIT), Fritz-Haber-Weg 2, 76131 Karlsruhe, Germany; [email protected] 2 Engler-Bunte-Institut, Chair of Water Chemistry and Water Technology, Karlsruhe Institute of Technology (KIT), Engler-Bunte-Ring 9, 76131 Karlsruhe, Germany; [email protected] (M.H.); [email protected] (H.H.) 3 Institute for Biological Interfaces, Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany * Correspondence: [email protected] Abstract: The biotechnological usage of carbon dioxide has become a relevant aim for future processes. Microbial electrosynthesis is a rather new technique to energize biological CO2 fixation with the advantage to establish a continuous process based on a cathodic biofilm that is supplied with renewable electrical energy as electron and energy source. In this study, the recently characterized cathodic biofilm forming microorganism Kyrpidia spormannii strain EA-1 was used in an adaptive laboratory evolution experiment to enhance its cathodic biofilm growth capabilities. At the end of the experiment, the adapted cathodic population exhibited an up to fourfold higher biofilm accumulation rate, as well as faster substratum coverage and a more uniform biofilm morphology compared to the progenitor strain. Genomic variant analysis revealed a genomically heterogeneous population with genetic variations occurring to various extends throughout the community. Via the conducted analysis we identified possible targets for future genetic engineering with the aim to further optimize Citation: Jung, T.; Hackbarth, M.; cathodic growth.