Human MLL/KMT2A Gene Exhibits a Second Breakpoint Cluster Region For
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis
International Journal of Molecular Sciences Review Aberrant Activity of Histone–Lysine N-Methyltransferase 2 (KMT2) Complexes in Oncogenesis Elzbieta Poreba 1,* , Krzysztof Lesniewicz 2 and Julia Durzynska 1,* 1 Institute of Experimental Biology, Faculty of Biology, Adam Mickiewicz University, ul. Uniwersytetu Pozna´nskiego6, 61-614 Pozna´n,Poland 2 Department of Molecular and Cellular Biology, Institute of Molecular Biology and Biotechnology, Faculty of Biology, Adam Mickiewicz University, ul. Uniwersytetu Pozna´nskiego6, 61-614 Pozna´n,Poland; [email protected] * Correspondence: [email protected] (E.P.); [email protected] (J.D.); Tel.: +48-61-829-5857 (E.P.) Received: 19 November 2020; Accepted: 6 December 2020; Published: 8 December 2020 Abstract: KMT2 (histone-lysine N-methyltransferase subclass 2) complexes methylate lysine 4 on the histone H3 tail at gene promoters and gene enhancers and, thus, control the process of gene transcription. These complexes not only play an essential role in normal development but have also been described as involved in the aberrant growth of tissues. KMT2 mutations resulting from the rearrangements of the KMT2A (MLL1) gene at 11q23 are associated with pediatric mixed-lineage leukemias, and recent studies demonstrate that KMT2 genes are frequently mutated in many types of human cancers. Moreover, other components of the KMT2 complexes have been reported to contribute to oncogenesis. This review summarizes the recent advances in our knowledge of the role of KMT2 complexes in cell transformation. In addition, it discusses the therapeutic targeting of different components of the KMT2 complexes. Keywords: histone–lysine N-methyltransferase 2; COMPASS; COMPASS-like; H3K4 methylation; oncogenesis; cancer; epigenetics; chromatin 1. -

Análise Integrativa De Perfis Transcricionais De Pacientes Com
UNIVERSIDADE DE SÃO PAULO FACULDADE DE MEDICINA DE RIBEIRÃO PRETO PROGRAMA DE PÓS-GRADUAÇÃO EM GENÉTICA ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Ribeirão Preto – 2012 ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas Tese apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo para obtenção do título de Doutor em Ciências. Área de Concentração: Genética Orientador: Prof. Dr. Eduardo Antonio Donadi Co-orientador: Prof. Dr. Geraldo A. S. Passos Ribeirão Preto – 2012 AUTORIZO A REPRODUÇÃO E DIVULGAÇÃO TOTAL OU PARCIAL DESTE TRABALHO, POR QUALQUER MEIO CONVENCIONAL OU ELETRÔNICO, PARA FINS DE ESTUDO E PESQUISA, DESDE QUE CITADA A FONTE. FICHA CATALOGRÁFICA Evangelista, Adriane Feijó Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. Ribeirão Preto, 2012 192p. Tese de Doutorado apresentada à Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. Área de Concentração: Genética. Orientador: Donadi, Eduardo Antonio Co-orientador: Passos, Geraldo A. 1. Expressão gênica – microarrays 2. Análise bioinformática por module maps 3. Diabetes mellitus tipo 1 4. Diabetes mellitus tipo 2 5. Diabetes mellitus gestacional FOLHA DE APROVAÇÃO ADRIANE FEIJÓ EVANGELISTA Análise integrativa de perfis transcricionais de pacientes com diabetes mellitus tipo 1, tipo 2 e gestacional, comparando-os com manifestações demográficas, clínicas, laboratoriais, fisiopatológicas e terapêuticas. -

Mutational Landscape and Clinical Outcome of Patients with De Novo Acute Myeloid Leukemia and Rearrangements Involving 11Q23/KMT2A
Mutational landscape and clinical outcome of patients with de novo acute myeloid leukemia and rearrangements involving 11q23/KMT2A Marius Billa,1,2, Krzysztof Mrózeka,1,2, Jessica Kohlschmidta,b, Ann-Kathrin Eisfelda,c, Christopher J. Walkera, Deedra Nicoleta,b, Dimitrios Papaioannoua, James S. Blachlya,c, Shelley Orwicka,c, Andrew J. Carrolld, Jonathan E. Kolitze, Bayard L. Powellf, Richard M. Stoneg, Albert de la Chapelleh,i,2, John C. Byrda,c, and Clara D. Bloomfielda,c aThe Ohio State University Comprehensive Cancer Center, Columbus, OH 43210; bAlliance for Clinical Trials in Oncology Statistics and Data Center, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210; cDivision of Hematology, Department of Internal Medicine, The Ohio State University Comprehensive Cancer Center, Columbus, OH 43210; dDepartment of Genetics, University of Alabama at Birmingham, Birmingham, AL 35294; eNorthwell Health Cancer Institute, Zucker School of Medicine at Hofstra/Northwell, Lake Success, NY 11042; fDepartment of Internal Medicine, Section on Hematology & Oncology, Wake Forest Baptist Comprehensive Cancer Center, Winston-Salem, NC 27157; gDepartment of Medical Oncology, Dana-Farber/Partners Cancer Care, Boston, MA 02215; hHuman Cancer Genetics Program, Comprehensive Cancer Center, The Ohio State University, Columbus, OH 43210; and iDepartment of Cancer Biology and Genetics, Comprehensive Cancer Center, The Ohio State University, Columbus, OH 43210 Contributed by Albert de la Chapelle, August 28, 2020 (sent for review July 17, 2020; reviewed by Anne Hagemeijer and Stefan Klaus Bohlander) Balanced rearrangements involving the KMT2A gene, located at patterns that include high expression of HOXA genes and thereby 11q23, are among the most frequent chromosome aberrations in contribute to leukemogenesis (14–16). -

A KMT2A-AFF1 Gene Regulatory Network Highlights the Role of Core Transcription Factors and Reveals the Regulatory Logic of Key Downstream Target Genes
Downloaded from genome.cshlp.org on October 7, 2021 - Published by Cold Spring Harbor Laboratory Press Research A KMT2A-AFF1 gene regulatory network highlights the role of core transcription factors and reveals the regulatory logic of key downstream target genes Joe R. Harman,1,7 Ross Thorne,1,7 Max Jamilly,2 Marta Tapia,1,8 Nicholas T. Crump,1 Siobhan Rice,1,3 Ryan Beveridge,1,4 Edward Morrissey,5 Marella F.T.R. de Bruijn,1 Irene Roberts,3,6 Anindita Roy,3,6 Tudor A. Fulga,2,9 and Thomas A. Milne1,6 1MRC Molecular Haematology Unit, MRC Weatherall Institute of Molecular Medicine, Radcliffe Department of Medicine, University of Oxford, Oxford, OX3 9DS, United Kingdom; 2MRC Weatherall Institute of Molecular Medicine, Radcliffe Department of Medicine, University of Oxford, Oxford, OX3 9DS, United Kingdom; 3MRC Molecular Haematology Unit, MRC Weatherall Institute of Molecular Medicine, Department of Paediatrics, University of Oxford, Oxford, OX3 9DS, United Kingdom; 4Virus Screening Facility, MRC Weatherall Institute of Molecular Medicine, John Radcliffe Hospital, University of Oxford, Oxford, OX3 9DS, United Kingdom; 5Center for Computational Biology, Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford OX3 9DS, United Kingdom; 6NIHR Oxford Biomedical Research Centre Haematology Theme, University of Oxford, Oxford, OX3 9DS, United Kingdom Regulatory interactions mediated by transcription factors (TFs) make up complex networks that control cellular behavior. Fully understanding these gene regulatory networks (GRNs) offers greater insight into the consequences of disease-causing perturbations than can be achieved by studying single TF binding events in isolation. Chromosomal translocations of the lysine methyltransferase 2A (KMT2A) gene produce KMT2A fusion proteins such as KMT2A-AFF1 (previously MLL-AF4), caus- ing poor prognosis acute lymphoblastic leukemias (ALLs) that sometimes relapse as acute myeloid leukemias (AMLs). -

Rare KMT2A-ELL and Novel ZNF56
CANCER GENOMICS & PROTEOMICS 18 : 121-131 (2021) doi:10.21873/cgp.20247 Rare KMT2A-ELL and Novel ZNF56-KMT2A Fusion Genes in Pediatric T-cell Acute Lymphoblastic Leukemia IOANNIS PANAGOPOULOS 1, KRISTIN ANDERSEN 1, MARTINE EILERT-OLSEN 1, ANNE GRO ROGNLIEN 2, MONICA CHENG MUNTHE-KAAS 2, FRANCESCA MICCI 1 and SVERRE HEIM 1,3 1Section for Cancer Cytogenetics, Institute for Cancer Genetics and Informatics, The Norwegian Radium Hospital, Oslo University Hospital, Oslo, Norway; 2Department of Pediatric Hematology and Oncology, Oslo University Hospital Rikshospitalet, Oslo, Norway; 3Institute of Clinical Medicine, Faculty of Medicine, University of Oslo, Oslo, Norway Abstract. Background/Aim: Previous reports have associated which could be distinguished by fluorescence in situ the KMT2A-ELL fusion gene, generated by t(11;19)(q23;p13.1), hybridization (FISH) (2, 3). Breakpoints within sub-band with acute myeloid leukemia (AML). We herein report a 19p13.3 have been found in both ALL (primarily in infants KMT2A-ELL and a novel ZNF56-KMT2A fusion genes in a and children) and AML. The translocation t(11;19)(q23;p13.3) pediatric T-lineage acute lymphoblastic leukemia (T-ALL). leads to fusion of the histone-lysine N-methyltransferase 2A Materials and Methods: Genetic investigations were performed (KMT2A; also known as myeloid/lymphoid or mixed lineage on bone marrow of a 13-year-old boy diagnosed with T-ALL. leukemia, MLL ) gene in 11q23 with the MLLT1 super Results: A KMT2A-ELL and a novel ZNF56-KMT2A fusion elongation complex subunit MLLT1 gene (also known as ENL, genes were generated on der(11)t(11;19)(q23;p13.1) and LTG19 , and YEATS1 ) in 19p13.3 generating a KMT2A-MLLT1 der(19)t(11;19)(q23;p13.1), respectively. -

UBASH3A Regulates the Synthesis and Dynamics of TCR–CD3 Complexes
UBASH3A Regulates the Synthesis and Dynamics of TCR−CD3 Complexes Yan Ge, Taylor K. Paisie, Sixue Chen and Patrick Concannon This information is current as of September 28, 2021. J Immunol published online 28 October 2019 http://www.jimmunol.org/content/early/2019/10/25/jimmun ol.1801338 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2019/10/25/jimmunol.180133 Material 8.DCSupplemental Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 28, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2019 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published October 28, 2019, doi:10.4049/jimmunol.1801338 The Journal of Immunology UBASH3A Regulates the Synthesis and Dynamics of TCR–CD3 Complexes Yan Ge,*,† Taylor K. Paisie,*,†,‡ Sixue Chen,†,‡,x,{,‖ and Patrick Concannon*,† The TCR–CD3 complex is a multicomponent membrane receptor, the expression of which is tightly regulated in thymo- cytes, as well as in mature T cells both at steady state and upon stimulation. -

Integrated Multimodal Genomic Analyses Reveal Novel Mechanisms of Glucocorticoid Resistance in Acute Lymphoblastic Leukemia
University of Tennessee Health Science Center UTHSC Digital Commons Theses and Dissertations (ETD) College of Graduate Health Sciences 4-2020 Integrated Multimodal Genomic Analyses Reveal Novel Mechanisms of Glucocorticoid Resistance in Acute Lymphoblastic Leukemia Robert J. Autry University of Tennessee Health Science Center Follow this and additional works at: https://dc.uthsc.edu/dissertations Part of the Medical Pharmacology Commons Recommended Citation Autry, Robert J. (https://orcid.org/0000-0002-6965-2942), "Integrated Multimodal Genomic Analyses Reveal Novel Mechanisms of Glucocorticoid Resistance in Acute Lymphoblastic Leukemia" (2020). Theses and Dissertations (ETD). Paper 516. http://dx.doi.org/10.21007/etd.cghs.2020.0501. This Dissertation is brought to you for free and open access by the College of Graduate Health Sciences at UTHSC Digital Commons. It has been accepted for inclusion in Theses and Dissertations (ETD) by an authorized administrator of UTHSC Digital Commons. For more information, please contact [email protected]. Integrated Multimodal Genomic Analyses Reveal Novel Mechanisms of Glucocorticoid Resistance in Acute Lymphoblastic Leukemia Abstract Acute lymphoblastic leukemia (ALL) is the most common childhood cancer. Much has been discovered in recent decades regarding ALL biology, and the outcome of patients with ALL has vastly improved, especially in pediatric ALL patients. Despite very promising overall cure rates, patients who relapse have a greatly decreased prognosis with survival rates ranging from 30-60%. These numbers stand to improve even further with new targeted therapies that seek to improve or maintain cure rates while reducing treatment related toxicities which affect patients both acutely and chronically. Glucocorticoids (GCs) are essential components of modern chemotherapeutic intervention for ALL. -

Ohnologs in the Human Genome Are Dosage Balanced and Frequently Associated with Disease
Ohnologs in the human genome are dosage balanced and frequently associated with disease Takashi Makino1 and Aoife McLysaght2 Smurfit Institute of Genetics, University of Dublin, Trinity College, Dublin 2, Ireland Edited by Michael Freeling, University of California, Berkeley, CA, and approved April 9, 2010 (received for review December 21, 2009) About 30% of protein-coding genes in the human genome are been duplicated by WGD, subsequent loss of individual genes related through two whole genome duplication (WGD) events. would result in a dosage imbalance due to insufficient gene Although WGD is often credited with great evolutionary impor- product, thus leading to biased retention of dosage-balanced tance, the processes governing the retention of these genes and ohnologs. In fact, evidence for preferential retention of dosage- their biological significance remain unclear. One increasingly pop- balanced genes after WGD is accumulating (4, 7, 11–20). Copy ular hypothesis is that dosage balance constraints are a major number variation [copy number polymorphism (CNV)] describes determinant of duplicate gene retention. We test this hypothesis population level polymorphism of small segmental duplications and show that WGD-duplicated genes (ohnologs) have rarely and is known to directly correlate with gene expression levels (21– experienced subsequent small-scale duplication (SSD) and are also 24). Thus, CNV of dosage-balanced genes is also expected to be refractory to copy number variation (CNV) in human populations deleterious. This model predicts that retained ohnologs should be and are thus likely to be sensitive to relative quantities (i.e., they are enriched for dosage-balanced genes that are resistant to sub- dosage-balanced). -

RUNX2 Regulates Leukemic Cell Metabolism and Chemotaxis in High-Risk T Cell Acute Lymphoblastic Leukemia
RUNX2 regulates leukemic cell metabolism and chemotaxis in high-risk T cell acute lymphoblastic leukemia Filip Matthijssens, … , Pieter Van Vlierberghe, Ksenia Matlawska-Wasowska J Clin Invest. 2021;131(6):e141566. https://doi.org/10.1172/JCI141566. Research Article Oncology Graphical abstract Find the latest version: https://jci.me/141566/pdf The Journal of Clinical Investigation RESEARCH ARTICLE RUNX2 regulates leukemic cell metabolism and chemotaxis in high-risk T cell acute lymphoblastic leukemia Filip Matthijssens,1,2 Nitesh D. Sharma,3,4 Monique Nysus,3,4 Christian K. Nickl,3,4 Huining Kang,4,5 Dominique R. Perez,4,6 Beatrice Lintermans,1,2 Wouter Van Loocke,1,2 Juliette Roels,1,2 Sofie Peirs,1,2 Lisa Demoen,1,2 Tim Pieters,1,2 Lindy Reunes,1,2 Tim Lammens,2,7 Barbara De Moerloose,2,7 Filip Van Nieuwerburgh,8 Dieter L. Deforce,8 Laurence C. Cheung,9,10 Rishi S. Kotecha,9,10 Martijn D.P. Risseeuw,2,11 Serge Van Calenbergh,2,11 Takeshi Takarada,12 Yukio Yoneda,13 Frederik W. van Delft,14 Richard B. Lock,15 Seth D. Merkley,5 Alexandre Chigaev,4,6 Larry A. Sklar,4,6 Charles G. Mullighan,16 Mignon L. Loh,17 Stuart S. Winter,18 Stephen P. Hunger,19 Steven Goossens,1,2,20 Eliseo F. Castillo,5 Wojciech Ornatowski,21 Pieter Van Vlierberghe,1,2 and Ksenia Matlawska-Wasowska3,4 1Department of Biomolecular Medicine, Ghent University, Ghent, Belgium. 2Cancer Research Institute Ghent (CRIG), Ghent, Belgium. 3Department of Pediatrics, Division of Hematology-Oncology, University of New Mexico Health Sciences Center, Albuquerque, New Mexico, USA. -

Histone Methylation Regulation in Neurodegenerative Disorders
International Journal of Molecular Sciences Review Histone Methylation Regulation in Neurodegenerative Disorders Balapal S. Basavarajappa 1,2,3,4,* and Shivakumar Subbanna 1 1 Division of Analytical Psychopharmacology, Nathan Kline Institute for Psychiatric Research, Orangeburg, NY 10962, USA; [email protected] 2 New York State Psychiatric Institute, New York, NY 10032, USA 3 Department of Psychiatry, College of Physicians & Surgeons, Columbia University, New York, NY 10032, USA 4 New York University Langone Medical Center, Department of Psychiatry, New York, NY 10016, USA * Correspondence: [email protected]; Tel.: +1-845-398-3234; Fax: +1-845-398-5451 Abstract: Advances achieved with molecular biology and genomics technologies have permitted investigators to discover epigenetic mechanisms, such as DNA methylation and histone posttransla- tional modifications, which are critical for gene expression in almost all tissues and in brain health and disease. These advances have influenced much interest in understanding the dysregulation of epigenetic mechanisms in neurodegenerative disorders. Although these disorders diverge in their fundamental causes and pathophysiology, several involve the dysregulation of histone methylation- mediated gene expression. Interestingly, epigenetic remodeling via histone methylation in specific brain regions has been suggested to play a critical function in the neurobiology of psychiatric disor- ders, including that related to neurodegenerative diseases. Prominently, epigenetic dysregulation currently brings considerable interest as an essential player in neurodegenerative disorders, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), Amyotrophic lateral sclerosis (ALS) and drugs of abuse, including alcohol abuse disorder, where it may facilitate connections between genetic and environmental risk factors or directly influence disease-specific Citation: Basavarajappa, B.S.; Subbanna, S. -
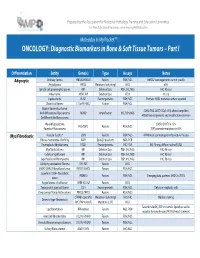
ONCOLOGY: Diagnostic Biomarkers in Bone & Soft Tissue Tumors – Part I
Prepared by the Association for Molecular Pathology Training and Education Committee For More Educational Resources: www.amp.org/AMPEducation Molecular In My PocketTM… ONCOLOGY: Diagnostic Biomarkers in Bone & Soft Tissue Tumors – Part I Differentiation Entity Gene(s) Type Assays Notes Adipocytic Ordinary lipoma HMGA2/HMGA1 Fusions FISH, NGS HMGA2 rearrangements are not specific Angiolipoma PRKD2 Mutations (activating) NGS 80% Spindle cell/pleomorphic lipoma RB1 Deletion/Loss FISH, IHC, NGS IHC: Rb loss Hibernoma MEN1, AIP Deletion/Loss FISH 11q13 Lipoblastoma PLAG1 Rearrangements FISH, NGS Partners: HAS2, numerous others reported Chondroid lipoma C11orf95-MKL2 Fusion FISH, NGS Atypical lipomatous tumor CDK4, FRS2, DDIT3 (12q14-15) often coamplified Well-differentiated liposarcoma MDM2 Amplification IHC, FISH, NGS HMGA2 rearrangements and amplification common Dedifferentiated liposarcoma Myxoid liposarcoma EWSR1-DDIT3 in <5% FUS-DDIT3 Fusions FISH, NGS Round cell liposarcoma TERT promoter mutations in 60% (Myo)Fibroblastic Nodular fasciitis* USP6 Fusions FISH, NGS MYH9 most common partner for nodular fascitis Fibrous hamartoma of infancy EGFR Exon 20 insertions NGS, PCR Desmoplastic fibroblastoma FOSL1 Rearrangements IHC, FISH IHC: Strong, diffuse nuclear FOSL1 Myofibroblastoma RB1 Deletion/Loss FISH, IHC, NGS IHC: Rb loss Cellular angiofibroma RB1 Deletion/Loss FISH, IHC, NGS IHC: Rb loss Superficial acral fibromyxoma RB1 Deletion/Loss FISH, IHC, NGS IHC: Rb loss Calcifying aponeurotic fibroma FN1-EGF Fusions NGS EWSR1-SMAD3 fibroblastic -

(NGS) for Primary Endocrine Resistance in Breast Cancer Patients
Int J Clin Exp Pathol 2018;11(11):5450-5458 www.ijcep.com /ISSN:1936-2625/IJCEP0084102 Original Article Impact of next-generation sequencing (NGS) for primary endocrine resistance in breast cancer patients Ruoyang Li1*, Tiantian Tang1*, Tianli Hui1, Zhenchuan Song1, Fugen Li2, Jingyu Li2, Jiajia Xu2 1Breast Center, Fourth Hospital of Hebei Medical University, Shijiazhuang, China; 2Institute of Precision Medicine, 3D Medicines Inc., Shanghai, China. *Equal contributors. Received August 15, 2018; Accepted September 22, 2018; Epub November 1, 2018; Published November 15, 2018 Abstract: Multiple mechanisms have been detected to account for the acquired resistance to endocrine therapies in breast cancer. In this study we retrospectively studied the mechanism of primary endocrine resistance in estrogen receptor positive (ER+) breast cancer patients by next-generation sequencing (NGS). Tumor specimens and matched blood samples were obtained from 24 ER+ breast cancer patients. Fifteen of them displayed endocrine resistance, including recurrence and/or metastases within 24 months from the beginning of endocrine therapy, and 9 pa- tients remained sensitive to endocrine therapy for more than 5 years. Genomic DNA of tumor tissue was extracted from formalin-fixed paraffin-embedded (FFPE) tumor tissue blocks. Genomic DNA of normal tissue was extracted from peripheral blood mononuclear cells (PBMC). Sequencing libraries for each sample were prepared, followed by target capturing for 372 genes that are frequently rearranged in cancers. Massive parallel