Metagenomics of a Microbial Mat
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Study on the Phototrophic Microbial Mat Communities of Sulphur Mountain Thermal Springs and Their Association with the Endangered, Endemic Snail Physella Johnsoni
A Study on the Phototrophic Microbial Mat Communities of Sulphur Mountain Thermal Springs and their Association with the Endangered, Endemic Snail Physella johnsoni By Michael Bilyj A thesis submitted to the Faculty of Graduate Studies in partial fulfillment of the requirements for the degree of Master of Science Department of Microbiology Faculty of Science University of Manitoba Winnipeg, Manitoba October 2011 © Copyright 2011, Michael A. Bilyj 1 Abstract The seasonal population fluctuation of anoxygenic phototrophs and the diversity of cyanobacteria at the Sulphur Mountain thermal springs of Banff, Canada were investigated and compared to the drastic population changes of the endangered snail Physella johnsoni. A new species and two strains of Rhodomicrobium were taxonomically characterized in addition to new species of Rhodobacter and Erythromicrobium. Major mat-forming organisms included Thiothrix-like species, oxygenic phototrophs of genera Spirulina, Oscillatoria, and Phormidium and purple nonsulfur bacteria Rhodobacter, Rhodopseudomonas and Rhodomicrobium. Aerobic anoxygenic phototrophs comprised upwards of 9.6 x 104 CFU/cm2 of mat or 18.9% of total aerobic heterotrophic bacterial isolates at certain sites, while maximal purple nonsulfur and purple sulfur bacteria were quantified at 3.2 x 105 and 2.0 x 106 CFU/cm2 of mat, respectively. Photosynthetic activity measurements revealed incredibly productive carbon fixation rates averaging 40.5 mg C/cm2/24 h. A temporal mismatch was observed for mat area and prokaryote-based organics to P. johnsoni population flux in a ―tracking inertia‖ manner. 2 Acknowledgements It is difficult to express sufficient gratitude to my supervisor Dr. Vladimir Yurkov for his unfaltering patience, generosity and motivation throughout this entire degree. -

Thermophilic Lithotrophy and Phototrophy in an Intertidal, Iron-Rich, Geothermal Spring 2 3 Lewis M
bioRxiv preprint doi: https://doi.org/10.1101/428698; this version posted September 27, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Thermophilic Lithotrophy and Phototrophy in an Intertidal, Iron-rich, Geothermal Spring 2 3 Lewis M. Ward1,2,3*, Airi Idei4, Mayuko Nakagawa2,5, Yuichiro Ueno2,5,6, Woodward W. 4 Fischer3, Shawn E. McGlynn2* 5 6 1. Department of Earth and Planetary Sciences, Harvard University, Cambridge, MA 02138 USA 7 2. Earth-Life Science Institute, Tokyo Institute of Technology, Meguro, Tokyo, 152-8550, Japan 8 3. Division of Geological and Planetary Sciences, California Institute of Technology, Pasadena, CA 9 91125 USA 10 4. Department of Biological Sciences, Tokyo Metropolitan University, Hachioji, Tokyo 192-0397, 11 Japan 12 5. Department of Earth and Planetary Sciences, Tokyo Institute of Technology, Meguro, Tokyo, 13 152-8551, Japan 14 6. Department of Subsurface Geobiological Analysis and Research, Japan Agency for Marine-Earth 15 Science and Technology, Natsushima-cho, Yokosuka 237-0061, Japan 16 Correspondence: [email protected] or [email protected] 17 18 Abstract 19 Hydrothermal systems, including terrestrial hot springs, contain diverse and systematic 20 arrays of geochemical conditions that vary over short spatial scales due to progressive interaction 21 between the reducing hydrothermal fluids, the oxygenated atmosphere, and in some cases 22 seawater. At Jinata Onsen, on Shikinejima Island, Japan, an intertidal, anoxic, iron- and 23 hydrogen-rich hot spring mixes with the oxygenated atmosphere and sulfate-rich seawater over 24 short spatial scales, creating an enormous range of redox environments over a distance ~10 m. -

The Vulnerability of Microbial Ecosystems in a Changing Climate: Potential Impact in Shark Bay
life Review The Vulnerability of Microbial Ecosystems in a Changing Climate: Potential Impact in Shark Bay Max Reinold 1,2, Hon Lun Wong 1,2, Fraser I. MacLeod 1,2, Julia Meltzer 1,2, April Thompson 1,2 and Brendan P. Burns 1,2,* 1 School of Biotechnology and Biomolecular Sciences, The University of New South Wales, Sydney 2052, Australia 2 Australian Centre for Astrobiology, The University of New South Wales, Sydney 2052, Australia * Correspondence: [email protected]; Tel.: +612-9385-3659; Fax: +612-9385-1591 Received: 30 July 2019; Accepted: 28 August 2019; Published: 2 September 2019 Abstract: The potential impact of climate change on eukaryotes, including humans, has been relatively well described. In contrast, the contribution and susceptibility of microorganisms to a changing climate have, until recently, received relatively less attention. In this review, the importance of microorganisms in the climate change discourse is highlighted. Microorganisms are responsible for approximately half of all primary production on earth, support all forms of macroscopic life whether directly or indirectly, and often persist in “extreme” environments where most other life are excluded. In short, microorganisms are the life support system of the biosphere and therefore must be included in decision making regarding climate change. Any effects climate change will have on microorganisms will inevitably impact higher eukaryotes and the activity of microbial communities in turn can contribute to or alleviate the severity of the changing climate. It is of vital importance that unique, fragile, microbial ecosystems are a focus of research efforts so that their resilience to extreme weather events and climate change are thoroughly understood and that conservation efforts can be implemented as a response. -

The Mysterious Microbial Mats
Submarine Ring of Fire 2012: NE Lau Basin Expedition The Mysterious Microbial Mats Focus Ecological role of microbial mats in hydrothermal vent ecosystems Grade Level 5-6 (Life Science) Focus Question What is the role of microbial mats in hydrothermal vent ecosystems? Learning Objectives n Students will plan an investigation using a model ecosystem to explain some of the components of an anaerobic ecosystem. n Students will construct explanations for the potential role of microbial mats in hydrothermal vent ecosystems. Materials For each student group: q Directions for setting up Winogradsky columns from http:// quest.arc.nasa.gov/projects/astrobiology/fieldwork/lessons/ Winogradsky_5_8.pdf q Two 2-liter plastic soda bottles q Black mud from a local river, lake, or estuary, approximately 2.5 l q 2.5 l of water from each mud/sand location used q 1 small bucket q 250 ml graduated cylinder or 1 cup measure q 1 paint stirrer or large spoon q 1 sheet of newspaper q 1 tablespoon powdered chalk q One hard boiled egg yolk or one tablespoon calcium sulfate q 1 set measuring spoons q Aluminum foil or plastic wrap and rubber band q Masking tape and markers for labeling columns q (Optional) Pencil sharpener for crushing chalk q (Optional) Mortar and pestle for making egg yolk powder q (Optional) 1 lamp with 40- to 60-watt light bulb q (Optional) Microscopes and materials for making wet mounts Image captions/credits on Page 2. Audio-Visual Materials q (Optional) Interactive whiteboard 1 www.oceanexplorer.noaa.gov Submarine Ring of Fire 2012: NE Lau -

“How to Make a Microbial Mat”
Science Lesson Plan: How to Make a Microbial Mat Brad Bebout, Allison Pfeifer, Erich Fleming, Adrienne Frisbee, Jan Winter NASA Ames Research Center Background Microbes have shaped the atmosphere and climate of Earth over geologic time. Microbes may be important in the search for life both within our solar system and on extrasolar planets. Some of the earliest and most widespread evidence of life on Earth were in the form of microbial mats. Microbial mats are complex, usually layered collections of microorganisms that can be observed in a number of aquatic ecosystems. They are complete ecosystems in miniature. In this activity, students create living versions of the microbial mats that have existed on earth for 3.5 billion years. Living microbial mats constructed in plastic boxes. Main Concepts Photosynthetic microbial mats are complex ecosystems that generally exhibit vertical gradients in light, and chemical compounds. These gradients are established by photosynthesis at the top of the mat and control the functioning of the intact ecosystem. Scientific Question What factors contribute to the vertical structure of microbial mats in the field? Objectives 1. The students will create their own microbial mat. 2. The microbial mat will be available for later experimentation and/or observation. Abstract of Lesson Microbial mats are defined as layered communities of microorganisms that are only a few millimeters thick. Many biogeochemical processes are conducted by the organisms in the mat to sustain life. For example, the mats perform photosynthesis, and respiration, and can acquire nitrogen through the process of nitrogen fixation. The mat community is a fully functioning ecosystem containing both producers and consumers or organic carbon. -

Microbial Lithification in Marine Stromatolites and Hypersaline Mats
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by RERO DOC Digital Library Published in Trends in Microbiology 13,9 : 429-438, 2005, 1 which should be used for any reference to this work Microbial lithification in marine stromatolites and hypersaline mats Christophe Dupraz1 and Pieter T. Visscher2 1Institut de Ge´ ologie, Universite´ de Neuchaˆ tel, Rue Emile-Argand 11, CP 2, CH-2007 Neuchaˆ tel, Switzerland 2Center for Integrative Geosciences, Department of Marine Sciences, University of Connecticut, 1080 Shennecossett Road, Groton, Connecticut, 06340, USA Lithification in microbial ecosystems occurs when pre- crucial role in regulating sedimentation and global bio- cipitation of minerals outweighs dissolution. Although geochemical cycles. the formation of various minerals can result from After the decline of stromatolites in the late Proterozoic microbial metabolism, carbonate precipitation is pos- (ca. 543 million years before present), microbially induced sibly the most important process that impacts global and/or controlled precipitation continued throughout the carbon cycling. Recent investigations have produced geological record as an active and essential player in most models for stromatolite formation in open marine aquatic ecosystems [9,10]. Although less abundant than in environments and lithification in shallow hypersaline the Precambrian, microbial precipitation is observed in a lakes, which could be highly relevant for interpreting the multitude of semi-confined (physically or chemically) -

Artificial Cold-Adapted Microbial Mats Cultured from Antarctic Lake Samples
AQUATIC MICROBIAL ECOLOGY Vol. 26: 127–138, 2001 Published December 5 Aquat Microb Ecol Artificial cold-adapted microbial mats cultured from Antarctic lake samples. 2. Short-term temperature effects on oxygen turn-over Olivier Pringault*, Evelyne Buffan-Dubau, Rutger de Wit** Station Marine d’Arcachon Laboratoire d’Océanographie Biologique CNRS-UMR 5805 Université Bordeaux-1, 2 rue du Prof. Jolyet, 33120 Arcachon, France ABSTRACT: Short-term temperature effects on oxygen turn-over in artificial cold-adapted microbial mats were experimentally studied using microsensor techniques. The artificial mats were cultured in benthic gradient chambers under opposing oxygen and sulphide gradients, and community meta- bolism was analysed after 8 mo of incubation at 5°C. Microbial mat samples from Lake Fryxell (Antarctica) were used as inocula. Both net and gross O2 productivity were maximal at 10°C, whereas gross O2 production was completely inhibited at 15°C. Thus, the microbial community exhibited a psychrophilic response. Nevertheless, the inhibition of oxygenic photosynthesis at 15°C might result from indirect effects of accumulation of H2S in the photic zone, which might induce the inhibition of photosystem II activity. The dynamics of the dark and light net metabolism of O2 were estimated from transient oxygen profiles. They were strongly affected by the temperature changes, exhibiting tem- perature characteristics well above those calculated from the steady-state approaches. This indicates that indirect effects of temperature probably occurred, i.e. (1) migration of microorganisms locally resulting in higher biomass, (2) an increase of the oxygen requirements for the oxidation of reduced compounds such as H2S, or both. -

Impacts of Seasonality and Nutrients on Microbial Mat Community Structure and Function
MARINE ECOLOGY PROGRESS SERIES Vol. 123: 207-216. 1995 Published July 20 Mar Ecol Prog Ser Impacts of seasonality and nutrients on microbial mat community structure and function J. Pinckney, H. W. Paerl, M. Fitzpatrick University of North Carolina at Chapel Hill, Institute of Marine Sciences, 3431 Arendell Street. Morehead City, North Carolina 28557, USA ABSTRACT. To understand the mechanisms responsible for seasonal fluctuations in growth and N2 fixation in intertidal microbial mat communties, we quantified seasonal changes in mat community composition, related these changes to die1 and seasonal N2 fixation rates, and evaluated community responses (growth, N2 fixation, composition) to long-term (22 d) nutrient addition bioassays. A tem- perate intertidal cyanobacterial mat community, located in coastal North Carolina, USA, was sampled at monthly intervals for 1 yr (1993-94) to determine changes in community composition. The abun- dances of major phototrophic groups were quantified based on the relative concentrations of taxa- specific photopigments (chlorophylls and carotenoids). The most abundant phototrophs were cyano- bacteria, diatoms, and photosynthetic bacteria. Mat blomass and community composition underwent marked changes on both monthly and seasonal scales and corresponded with seasonal shifts in the dlel patterns of N2 fixation. Diatom biomass increased during periods of low N2 fixation. Nutnent (nitrate and phosphate) addition bioassays indicated that both cyanobacterial and diatom growth were N limited. Cyanobacteria were able to circumvent N llmltation by N2 fixation. The addition of high con- centrations of N (100 ,pM NaNO,) in combination with P (100 ~JMNaH2P04) resulted In an Increase (163'Y") in the relative abundance of diatoms The addition of P alone more than doubled N2 fixation rates and cyanobacterial abundance increased (+34'h) relative to diatoms. -

Biodiversity of the Microbial Mat of the Garga Hot Spring
Rozanov et al. BMC Evolutionary Biology 2017, 17(Suppl 2):254 DOI 10.1186/s12862-017-1106-9 RESEARCH Open Access Biodiversity of the microbial mat of the Garga hot spring Alexey Sergeevich Rozanov1*, Alla Victorovna Bryanskaya1, Timofey Vladimirovich Ivanisenko1,2, Tatyana Konstantinovna Malup1 and Sergey Evgenievich Peltek1 From Belyaev Conference Novosibirsk, Russia. 07-10 August 2017 Abstract Background: Microbial mats are a good model system for ecological and evolutionary analysis of microbial communities. There are more than 20 alkaline hot springs on the banks of the Barguzin river inflows. Water temperature reaches 75 °C and pH is usually 8.0–9.0. The formation of microbial mats is observed in all hot springs. Microbial communities of hot springs of the Baikal rift zone are poorly studied. Garga is the biggest hot spring in this area. Results: In this study, we investigated bacterial and archaeal diversity of the Garga hot spring (Baikal rift zone, Russia) using 16S rRNA metagenomic sequencing. We studied two types of microbial communities: (i) small white biofilms on rocks in the points with the highest temperature (75 °C) and (ii) continuous thick phototrophic microbial mats observed at temperatures below 70 °C. Archaea (mainly Crenarchaeota; 19.8% of the total sequences) were detected only in the small biofilms. The high abundance of Archaea in the sample from hot springs of the Baikal rift zone supplemented our knowledge of the distribution of Archaea. Most archaeal sequences had low similarity to known Archaea. In the microbial mats, primary products were formed by cyanobacteria of the genus Leptolyngbya. Heterotrophic microorganisms were mostly represented by Actinobacteria and Proteobacteria in all studied samples of the microbial mats. -

Unravelling Hidden Metabolic Capabilities in Microbial Mat
& Bioch ial em b ic ro a c l i T M e f c Mohammad Al-Najjar, J Microb Biochem Technol 2016, 8:1 h o Journal of n l o a n l o r DOI: 10.4172/1948-5948.1000256 g u y o J ISSN: 1948-5948 Microbial & Biochemical Technology Short Communication Open Access Unravelling Hidden Metabolic Capabilities in Microbial Mat Ecosystems by Combining In Situ Measurements and Omic Approaches Mohammad AA Al-Najjar* Red Sea Research Center, KAUST, Thuwal, Saudi Arabia Abstract Microbial mats are unique ecosystems that harbor diversified metabolic capabilities raging from photosynthesis in the upper layer to fermentation and organic material degradation accompanied with methane production in the deepest layer. Microorganisms in a microbial mat are successful to flourish in extreme environments because they can shift their metabolism in response to the changes in their environment. These metabolic flexibility and capabilities of the microorganisms are underestimated because most of the studies so far have focused on measuring changing in the behavior of the inhabiting microorganisms, or on certain biochemical indicators, or on the shift of microbial community structure. However, to be able to broaden our understanding of the metabolic capabilities, an intensive study of the underlying molecular mechanisms should be carried out. Therefore, the best approach to be adopted is by combining in situ measurements with omics analysis (i.e., matatranscriptomic and/or metaproteomic) in response to changes in the environmental conditions. Microbial mats are complete ecosystems on a millimeter scale very interesting as they have revealed new metabolic possibilities for that harbor diversified metabolic processes [1-5]. -

Microbial Mat Ecosystems: Structure Types, Functional Diversity, and Biotechnological Application
Electronic Journal of Biotechnology 31 (2018) 48–56 Contents lists available at ScienceDirect Electronic Journal of Biotechnology Review Microbial mat ecosystems: Structure types, functional diversity, and biotechnological application Cristina M. Prieto-Barajas a, Eduardo Valencia-Cantero b, Gustavo Santoyo a,⁎ a Laboratorio de Diversidad Genómica, Instituto de Investigaciones Químico-Biológicas, Universidad Michoacana de San Nicolás de Hidalgo, Edificio A1, Ciudad Universitaria, C.P. 58063, Morelia, Michoacán, Mexico b Laboratorio de Ecología Microbiana, Instituto de Investigaciones Químico-Biológicas, Universidad Michoacana de San Nicolás de Hidalgo, Edificio A1, Ciudad Universitaria, C.P. 58063, Morelia, Michoacán, Mexico article info abstract Article history: Microbial mats are horizontally stratified microbial communities, exhibiting a structure defined by Received 10 July 2017 physiochemical gradients, which models microbial diversity, physiological activities, and their dynamics as a Accepted 10 November 2017 whole system. These ecosystems are commonly associated with aquatic habitats, including hot springs, Available online 21 November 2017 hypersaline ponds, and intertidal coastal zones and oligotrophic environments, all of them harbour phototrophic mats and other environments such as acidic hot springs or acid mine drainage harbour Keywords: non-photosynthetic mats. This review analyses the complex structure, diversity, and interactions between the Acid microbial mats Biofilm microorganisms that form the framework of different types of microbial mats located around the globe. Coastal mats Furthermore, the many tools that allow studying microbial mats in depth and their potential biotechnological Extreme environments applications are discussed. Hot springs Hypersaline mats © 2017 PontificiaUniversidadCatólicadeValparaíso.Productionand hosting by Elsevier B.V. All rights reserved. Microbial biotechnology This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/). -
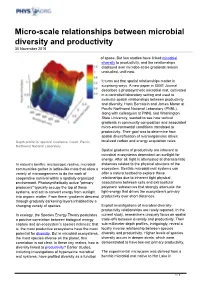
Micro-Scale Relationships Between Microbial Diversity and Productivity 30 November 2016
Micro-scale relationships between microbial diversity and productivity 30 November 2016 of space. But few studies have linked microbial diversity to productivity, and the relationships displayed over microbe-scale gradients remain unstudied, until now. It turns out that spatial relationships matter in surprising ways. A new paper in ISME Journal describes a photosynthetic microbial mat, cultivated in a controlled laboratory setting and used to evaluate spatial relationships between productivity and diversity. Hans Bernstein and James Moran at Pacific Northwest National Laboratory (PNNL), along with colleagues at PNNL and Washington State University, wanted to see how vertical gradients in community composition and associated micro-environmental conditions translated to productivity. Their goal was to determine how spatial diversification of microorganisms drives Depth profile for spectral irradiance. Credit: Pacific localized carbon and energy acquisition rates. Northwest National Laboratory Spatial gradients of productivity are inherent to microbial ecosystems dependent on sunlight for energy. After all, light is attenuated at characteristic In nature's benthic microscopic realms, microbial distances related to the physical structure of the communities gather in lattice-like mats that allow a ecosystem. Benthic microbial mat systems can variety of microorganisms to do the work of offer a natural testbed to explore these cooperative survival within a spatially organized relationships due to inherent tight physical environment. Photosynthetically active "primary associations between cells and extracellular producers" typically occupy the top of these polymeric substances that strongly attenuate the systems, and act to convert energy from sunlight light-energy that drives the ecosystem's primary into organic matter. From there, gradients descend productivity over short distances.