The Glucocorticoid-Mediated Dynamics of Genome Architecture
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Anti-ARL4A Antibody (ARG41291)
Product datasheet [email protected] ARG41291 Package: 100 μl anti-ARL4A antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes ARL4A Tested Reactivity Hu, Ms, Rat Tested Application ICC/IF, IHC-P Host Rabbit Clonality Polyclonal Isotype IgG Target Name ARL4A Antigen Species Human Immunogen Recombinant fusion protein corresponding to aa. 121-200 of Human ARL4A (NP_001032241.1). Conjugation Un-conjugated Alternate Names ARL4; ADP-ribosylation factor-like protein 4A Application Instructions Application table Application Dilution ICC/IF 1:50 - 1:200 IHC-P 1:50 - 1:200 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 23 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.3), 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. www.arigobio.com 1/2 Bioinformation Gene Symbol ARL4A Gene Full Name ADP-ribosylation factor-like 4A Background ADP-ribosylation factor-like 4A is a member of the ADP-ribosylation factor family of GTP-binding proteins. ARL4A is similar to ARL4C and ARL4D and each has a nuclear localization signal and an unusually high guaninine nucleotide exchange rate. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -
![TCEAL1 Mouse Monoclonal Antibody [Clone ID: OTI3G9] Product Data](https://docslib.b-cdn.net/cover/1170/tceal1-mouse-monoclonal-antibody-clone-id-oti3g9-product-data-731170.webp)
TCEAL1 Mouse Monoclonal Antibody [Clone ID: OTI3G9] Product Data
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for CF807707 TCEAL1 Mouse Monoclonal Antibody [Clone ID: OTI3G9] Product data: Product Type: Primary Antibodies Clone Name: OTI3G9 Applications: WB Recommended Dilution: WB 1:2000 Reactivity: Human Host: Mouse Isotype: IgG1 Clonality: Monoclonal Immunogen: Full length human recombinant protein of human TCEAL1 (NP_004771) produced in E.coli. Formulation: Lyophilized powder (original buffer 1X PBS, pH 7.3, 8% trehalose) Reconstitution Method: For reconstitution, we recommend adding 100uL distilled water to a final antibody concentration of about 1 mg/mL. To use this carrier-free antibody for conjugation experiment, we strongly recommend performing another round of desalting process. (OriGene recommends Zeba Spin Desalting Columns, 7KMWCO from Thermo Scientific) Purification: Purified from mouse ascites fluids or tissue culture supernatant by affinity chromatography (protein A/G) Conjugation: Unconjugated Storage: Store at -20°C as received. Stability: Stable for 12 months from date of receipt. Predicted Protein Size: 18.5 kDa Gene Name: Homo sapiens transcription elongation factor A like 1 (TCEAL1), transcript variant 1, mRNA. Database Link: NP_004771 Entrez Gene 9338 Human Q15170 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 TCEAL1 Mouse Monoclonal Antibody [Clone ID: OTI3G9] – CF807707 Background: This gene encodes a member of the transcription elongation factor A (SII)-like (TCEAL) gene family. -

Functional Annotation of Novel Lineage-Specific Genes Using Co-Expression and Promoter Analysis Charu G Kumar1, Robin E Everts1,3, Juan J Loor1, Harris a Lewin1,2*
Kumar et al. BMC Genomics 2010, 11:161 http://www.biomedcentral.com/1471-2164/11/161 RESEARCH ARTICLE Open Access Functional annotation of novel lineage-specific genes using co-expression and promoter analysis Charu G Kumar1, Robin E Everts1,3, Juan J Loor1, Harris A Lewin1,2* Abstract Background: The diversity of placental architectures within and among mammalian orders is believed to be the result of adaptive evolution. Although, the genetic basis for these differences is unknown, some may arise from rapidly diverging and lineage-specific genes. Previously, we identified 91 novel lineage-specific transcripts (LSTs) from a cow term-placenta cDNA library, which are excellent candidates for adaptive placental functions acquired by the ruminant lineage. The aim of the present study was to infer functions of previously uncharacterized lineage- specific genes (LSGs) using co-expression, promoter, pathway and network analysis. Results: Clusters of co-expressed genes preferentially expressed in liver, placenta and thymus were found using 49 previously uncharacterized LSTs as seeds. Over-represented composite transcription factor binding sites (TFBS) in promoters of clustered LSGs and known genes were then identified computationally. Functions were inferred for nine previously uncharacterized LSGs using co-expression analysis and pathway analysis tools. Our results predict that these LSGs may function in cell signaling, glycerophospholipid/fatty acid metabolism, protein trafficking, regulatory processes in the nucleus, and processes that initiate parturition and immune system development. Conclusions: The placenta is a rich source of lineage-specific genes that function in the adaptive evolution of placental architecture and functions. We have shown that co-expression, promoter, and gene network analyses are useful methods to infer functions of LSGs with heretofore unknown functions. -

Snp Associations with Tuberculosis Susceptibility in a Ugandan
SNP ASSOCIATIONS WITH TUBERCULOSIS SUSCEPTIBILITY IN A UGANDAN HOUSEHOLD CONTACT STUDY by ALLISON REES BAKER Submitted in partial fulfillment of the requirements For the degree of Master of Science Thesis Advisor: Dr. Catherine M. Stein Department of Epidemiology and Biostatistics CASE WESTERN RESERVE UNIVERSITY August, 2010 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of ______________________________________________________ candidate for the ________________________________degree *. (signed)_______________________________________________ (chair of the committee) ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. Table of Contents Table of Contents...............................................................................................................iii List of Tables ..................................................................................................................... iv Acknowledgements............................................................................................................. v List of Commonly Used Abbreviations ............................................................................. vi Chapter 1: Literature -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

Gait Abnormalities and Progressive Myelin Degeneration in a New Murine Model of Pelizaeus-Merzbacher Disease with Tandem Genomic Duplication
11788 • The Journal of Neuroscience, July 17, 2013 • 33(29):11788–11799 Neurobiology of Disease Gait Abnormalities and Progressive Myelin Degeneration in a New Murine Model of Pelizaeus-Merzbacher Disease with Tandem Genomic Duplication Kristi Clark,1,2 Lauren Sakowski,1,2 Karen Sperle,1 Linda Banser,1 Carlisle P. Landel,3 Denise A. Bessert,4 Robert P. Skoff,4 and Grace M. Hobson1,2,5 1Nemours Biomedical Research, Alfred I. duPont Hospital for Children, Wilmington, Delaware 19803, 2University of Delaware, Department of Biology, Newark, Delaware 19716, 3Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, Pennsylvania 19107, 4Wayne State University, Department of Anatomy and Cell Biology, Detroit, Michigan 48201, and 5Jefferson Medical College, Thomas Jefferson University, Philadelphia, Pennsylvania 19107 Pelizaeus-Merzbacher disease (PMD) is a hypomyelinating leukodystrophy caused by mutations of the proteolipid protein 1 gene (PLP1), which is located on the X chromosome and encodes the most abundant protein of myelin in the central nervous sytem. Approximately 60% of PMD cases result from genomic duplications of a region of the X chromosome that includes the entire PLP1 gene. The duplications are typically in a head-to-tail arrangement, and they vary in size and gene content. Although rodent models with extra copies of Plp1 have been developed, none contains an actual genomic rearrangement that resembles those found in PMD patients. We used mutagenic insertion chromosome engineering resources to generate the Plp1dup mouse model by introducing an X chromosome duplication in the mouse genome that contains Plp1 and five neighboring genes that are also commonly duplicated in PMD patients. The Plp1dup mice display progressive gait abnormalities compared with wild-type littermates. -
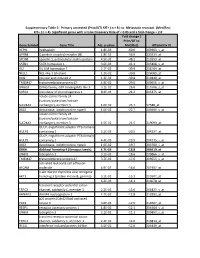
Gene Symbol Gene Title Adj. P-‐Value Fold Change ( Prim/UT to Met
Supplementary Table 2: Primary untreated (Prim/UT) KIT+ ( n = 8 ) vs. Metastatic resistant (Met/Res) KIT+ ( n = 4). Significant genes with a False Discovery Rate of < 0.05 and a fold change > 2.0 Fold change ( Prim/UT to Gene Symbol Gene Title Adj. p-value Met/Res) Affymetrix ID HEPH hephaestin 1.8E-03 -60.6 203903_s_at GPR88 G protein-coupled receptor 88 1.8E-02 -56.0 220313_at SPON1 spondin 1, extracellular matrix protein 4.5E-02 -48.2 213993_at SATB1 SATB homeobox 1 3.6E-03 -41.1 203408_s_at ISL1 ISL LIM homeobox 1 2.7E-02 -39.9 206104_at NELL1 NEL-like 1 (chicken) 1.2E-02 -39.8 206089_at RAI2 retinoic acid induced 2 1.3E-02 -30.8 219440_at TMEM47 transmembrane protein 47 4.9E-02 -29.0 209656_s_at DIRAS3 DIRAS family, GTP-binding RAS-like 3 3.1E-02 -26.8 215506_s_at SCRG1 stimulator of chondrogenesis 1 8.3E-03 -24.2 205475_at solute carrier family 24 (sodium/potassium/calcium SLC24A3 exchanger), member 3 1.6E-02 -23.7 57588_at DIO2 deiodinase, iodothyronine, type II 1.0E-02 -22.7 203699_s_at solute carrier family 24 (sodium/potassium/calcium SLC24A3 exchanger), member 3 1.5E-02 -21.5 219090_at GULP, engulfment adaptor PTB domain GULP1 containing 1 5.2E-03 -20.5 204237_at GULP, engulfment adaptor PTB domain GULP1 containing 1 4.4E-03 -19.9 204235_s_at DIO2 deiodinase, iodothyronine, type II 1.0E-02 -19.7 203700_s_at DKK4 dickkopf homolog 4 (Xenopus laevis) 4.7E-03 -18.8 206619_at LPHN3 latrophilin 3 3.1E-02 -18.6 209866_s_at TMEM47 transmembrane protein 47 3.7E-02 -17.6 209655_s_at activated leukocyte cell adhesion ALCAM molecule 4.9E-02 -

Modeling Genomic Diversity and Tumor Dependency in Malignant Melanoma
Research Article Modeling Genomic Diversity and Tumor Dependency in Malignant Melanoma William M. Lin,1,3,5 Alissa C. Baker,1,3 Rameen Beroukhim,1,3,5 Wendy Winckler,1,3,5 Whei Feng,1,3,5 Jennifer M. Marmion,7 Elisabeth Laine,8 Heidi Greulich,1,3,5 Hsiuyi Tseng,1,3 Casey Gates,5 F. Stephen Hodi,1 Glenn Dranoff,1 William R. Sellers,1,6 Roman K. Thomas,9,10 Matthew Meyerson,1,3,4,5 Todd R. Golub,2,3,5 Reinhard Dummer,8 Meenhard Herlyn,7 Gad Getz,3,5 and Levi A. Garraway1,3,5 Departments of 1Medical Oncology and 2Pediatric Oncology and 3Center for Cancer Genome Discovery, Dana-Farber Cancer Institute, Harvard Medical School; 4Department of Pathology, Harvard Medical School, Boston, Massachusetts; 5The Broad Institute of M.I.T. and Harvard; 6Novartis Institutes for Biomedical Research, Cambridge, Massachusetts; 7Cancer Biology Division, Wistar Institute, Philadelphia, Pennsylvania; 8Department of Dermatology, University of Zurich Hospital, Zu¨rich, Switzerland; 9Max Planck Institute for Neurological Research with Klaus Joachim Zulch Laboratories of the Max Planck Society and the Medical Faculty of the University of Cologne; and 10Center for Integrated Oncology and Department I for Internal Medicine, University of Cologne, Cologne, Germany Abstract tumorigenesis have been derived from functional studies involving The classification of human tumors based on molecular cultured human cancer cells (e.g., established cell lines, short-term cultures, etc.). Despite their limitations, cancer cell line collections criteria offers tremendous clinical potential; however, dis- cerning critical and ‘‘druggable’’ effectors on a large scale will whose genetic alterations reflect their primary tumor counterparts also require robust experimental models reflective of tumor should provide malleable proxies that facilitate mechanistic genomic diversity. -

Identifying Genetic Signatures of Recent Local Adaptations in People from Ibiza
Master Thesis UPPSALA UNIVERSITET Identifying genetic signatures of recent local adaptations in people from Ibiza Submitted for the degree of Master of Science Diego Alejandro Londoño Correa Supervised by Elena Bosch (Institute of Evolutionary Biology, CSIC-UPF, Barcelona) Carina Schlebusch (internal supervisor at Uppsala University) March 2021 1 Table of Contents ABSTRACT .................................................................................................................................................. 4 INTRODUCTION ......................................................................................................................................... 5 MATERIALS AND METHODS ....................................................................................................................... 8 Data ....................................................................................................................................................... 8 Quality control ....................................................................................................................................... 8 Pruning linked variants, PCA and Admixture ......................................................................................... 9 Selection Tests ....................................................................................................................................... 9 Control for iHS signals in Iberia ...........................................................................................................