Functional Annotation of Novel Lineage-Specific Genes Using Co-Expression and Promoter Analysis Charu G Kumar1, Robin E Everts1,3, Juan J Loor1, Harris a Lewin1,2*
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Anti-ARL4A Antibody (ARG41291)
Product datasheet [email protected] ARG41291 Package: 100 μl anti-ARL4A antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes ARL4A Tested Reactivity Hu, Ms, Rat Tested Application ICC/IF, IHC-P Host Rabbit Clonality Polyclonal Isotype IgG Target Name ARL4A Antigen Species Human Immunogen Recombinant fusion protein corresponding to aa. 121-200 of Human ARL4A (NP_001032241.1). Conjugation Un-conjugated Alternate Names ARL4; ADP-ribosylation factor-like protein 4A Application Instructions Application table Application Dilution ICC/IF 1:50 - 1:200 IHC-P 1:50 - 1:200 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 23 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.3), 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. www.arigobio.com 1/2 Bioinformation Gene Symbol ARL4A Gene Full Name ADP-ribosylation factor-like 4A Background ADP-ribosylation factor-like 4A is a member of the ADP-ribosylation factor family of GTP-binding proteins. ARL4A is similar to ARL4C and ARL4D and each has a nuclear localization signal and an unusually high guaninine nucleotide exchange rate. -

Mapping of Leptin and Its Syntenic Genes to Chicken Chromosome
Edinburgh Research Explorer Mapping of leptin and its syntenic genes to chicken chromosome 1p Citation for published version: Seroussi, E, Pitel, F, Leroux, S, Morisson, M, Bornelöv, S, Miyara, S, Yosefi, S, Cogburn, LA, Burt, DW, Anderson, L & Friedman-Einat, M 2017, 'Mapping of leptin and its syntenic genes to chicken chromosome 1p', BMC Genetics, vol. 18, no. 1, pp. 77. https://doi.org/10.1186/s12863-017-0543-1 Digital Object Identifier (DOI): 10.1186/s12863-017-0543-1 Link: Link to publication record in Edinburgh Research Explorer Document Version: Publisher's PDF, also known as Version of record Published In: BMC Genetics General rights Copyright for the publications made accessible via the Edinburgh Research Explorer is retained by the author(s) and / or other copyright owners and it is a condition of accessing these publications that users recognise and abide by the legal requirements associated with these rights. Take down policy The University of Edinburgh has made every reasonable effort to ensure that Edinburgh Research Explorer content complies with UK legislation. If you believe that the public display of this file breaches copyright please contact [email protected] providing details, and we will remove access to the work immediately and investigate your claim. Download date: 07. Oct. 2021 Seroussi et al. BMC Genetics (2017) 18:77 DOI 10.1186/s12863-017-0543-1 RESEARCHARTICLE Open Access Mapping of leptin and its syntenic genes to chicken chromosome 1p Eyal Seroussi1*, Frédérique Pitel2, Sophie Leroux2, Mireille Morisson2, Susanne Bornelöv3, Shoval Miyara1, Sara Yosefi1, Larry A. Cogburn4, David W. Burt5, Leif Anderson3,6,7 and Miriam Friedman-Einat1* Abstract Background: Misidentification of the chicken leptin gene has hampered research of leptin signaling in this species for almost two decades. -

Whole-Genome Rnai Screen Highlights Components of the Endoplasmic Reticulum/Golgi As a Source of Resistance to Immunotoxin-Media
Whole-genome RNAi screen highlights components of PNAS PLUS the endoplasmic reticulum/Golgi as a source of resistance to immunotoxin-mediated cytotoxicity Matteo Pasettoa,1,2, Antonella Antignania,1, Pinar Ormanoglub, Eugen Buehlerb, Rajarshi Guhab, Ira Pastana,3, Scott E. Martinb, and David J. FitzGeralda,3 aLaboratory of Molecular Biology, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892-4264; and bDivision of Preclinical Innovation, National Center for Advancing Translational Sciences, National Institutes of Health, Rockville, MD 20850 Contributed by Ira Pastan, February 2, 2015 (sent for review October 6, 2014; reviewed by Arthur Frankel and Wayne I. Lencer) Immunotoxins (antibody–toxin fusion proteins) target surface EF2 no longer functions at the elongation step of protein trans- antigens on cancer cells and kill these cells via toxin-mediated in- lation. Death results from a combination of events: the loss of hibition of protein synthesis. To identify genes controlling this short-lived survival proteins (e.g., Mcl1), triggering apoptosis, process, an RNAi whole-genome screen (∼22,000 genes at three and stress responses that cannot be executed because protein siRNAs per gene) was conducted via monitoring the cytotoxicity of translation is shut down (21). Regarding the constituents of the the mesothelin-directed immunotoxin SS1P. SS1P, a Pseudomonas pathway responsible for transporting the immunotoxin from the exotoxin-based immunotoxin, was chosen because it is now in surface to the cytosol, there are only a handful of experimentally clinical trials and has produced objective tumor regressions in established components. These proteins include the surface patients. High and low concentrations of SS1P were chosen to target itself, the protease furin, and KDELR2 (discussed below). -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Snp Associations with Tuberculosis Susceptibility in a Ugandan
SNP ASSOCIATIONS WITH TUBERCULOSIS SUSCEPTIBILITY IN A UGANDAN HOUSEHOLD CONTACT STUDY by ALLISON REES BAKER Submitted in partial fulfillment of the requirements For the degree of Master of Science Thesis Advisor: Dr. Catherine M. Stein Department of Epidemiology and Biostatistics CASE WESTERN RESERVE UNIVERSITY August, 2010 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of ______________________________________________________ candidate for the ________________________________degree *. (signed)_______________________________________________ (chair of the committee) ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. Table of Contents Table of Contents...............................................................................................................iii List of Tables ..................................................................................................................... iv Acknowledgements............................................................................................................. v List of Commonly Used Abbreviations ............................................................................. vi Chapter 1: Literature -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -
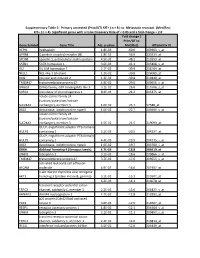
Gene Symbol Gene Title Adj. P-‐Value Fold Change ( Prim/UT to Met
Supplementary Table 2: Primary untreated (Prim/UT) KIT+ ( n = 8 ) vs. Metastatic resistant (Met/Res) KIT+ ( n = 4). Significant genes with a False Discovery Rate of < 0.05 and a fold change > 2.0 Fold change ( Prim/UT to Gene Symbol Gene Title Adj. p-value Met/Res) Affymetrix ID HEPH hephaestin 1.8E-03 -60.6 203903_s_at GPR88 G protein-coupled receptor 88 1.8E-02 -56.0 220313_at SPON1 spondin 1, extracellular matrix protein 4.5E-02 -48.2 213993_at SATB1 SATB homeobox 1 3.6E-03 -41.1 203408_s_at ISL1 ISL LIM homeobox 1 2.7E-02 -39.9 206104_at NELL1 NEL-like 1 (chicken) 1.2E-02 -39.8 206089_at RAI2 retinoic acid induced 2 1.3E-02 -30.8 219440_at TMEM47 transmembrane protein 47 4.9E-02 -29.0 209656_s_at DIRAS3 DIRAS family, GTP-binding RAS-like 3 3.1E-02 -26.8 215506_s_at SCRG1 stimulator of chondrogenesis 1 8.3E-03 -24.2 205475_at solute carrier family 24 (sodium/potassium/calcium SLC24A3 exchanger), member 3 1.6E-02 -23.7 57588_at DIO2 deiodinase, iodothyronine, type II 1.0E-02 -22.7 203699_s_at solute carrier family 24 (sodium/potassium/calcium SLC24A3 exchanger), member 3 1.5E-02 -21.5 219090_at GULP, engulfment adaptor PTB domain GULP1 containing 1 5.2E-03 -20.5 204237_at GULP, engulfment adaptor PTB domain GULP1 containing 1 4.4E-03 -19.9 204235_s_at DIO2 deiodinase, iodothyronine, type II 1.0E-02 -19.7 203700_s_at DKK4 dickkopf homolog 4 (Xenopus laevis) 4.7E-03 -18.8 206619_at LPHN3 latrophilin 3 3.1E-02 -18.6 209866_s_at TMEM47 transmembrane protein 47 3.7E-02 -17.6 209655_s_at activated leukocyte cell adhesion ALCAM molecule 4.9E-02 -

Modeling Genomic Diversity and Tumor Dependency in Malignant Melanoma
Research Article Modeling Genomic Diversity and Tumor Dependency in Malignant Melanoma William M. Lin,1,3,5 Alissa C. Baker,1,3 Rameen Beroukhim,1,3,5 Wendy Winckler,1,3,5 Whei Feng,1,3,5 Jennifer M. Marmion,7 Elisabeth Laine,8 Heidi Greulich,1,3,5 Hsiuyi Tseng,1,3 Casey Gates,5 F. Stephen Hodi,1 Glenn Dranoff,1 William R. Sellers,1,6 Roman K. Thomas,9,10 Matthew Meyerson,1,3,4,5 Todd R. Golub,2,3,5 Reinhard Dummer,8 Meenhard Herlyn,7 Gad Getz,3,5 and Levi A. Garraway1,3,5 Departments of 1Medical Oncology and 2Pediatric Oncology and 3Center for Cancer Genome Discovery, Dana-Farber Cancer Institute, Harvard Medical School; 4Department of Pathology, Harvard Medical School, Boston, Massachusetts; 5The Broad Institute of M.I.T. and Harvard; 6Novartis Institutes for Biomedical Research, Cambridge, Massachusetts; 7Cancer Biology Division, Wistar Institute, Philadelphia, Pennsylvania; 8Department of Dermatology, University of Zurich Hospital, Zu¨rich, Switzerland; 9Max Planck Institute for Neurological Research with Klaus Joachim Zulch Laboratories of the Max Planck Society and the Medical Faculty of the University of Cologne; and 10Center for Integrated Oncology and Department I for Internal Medicine, University of Cologne, Cologne, Germany Abstract tumorigenesis have been derived from functional studies involving The classification of human tumors based on molecular cultured human cancer cells (e.g., established cell lines, short-term cultures, etc.). Despite their limitations, cancer cell line collections criteria offers tremendous clinical potential; however, dis- cerning critical and ‘‘druggable’’ effectors on a large scale will whose genetic alterations reflect their primary tumor counterparts also require robust experimental models reflective of tumor should provide malleable proxies that facilitate mechanistic genomic diversity. -

Identifying Genetic Signatures of Recent Local Adaptations in People from Ibiza
Master Thesis UPPSALA UNIVERSITET Identifying genetic signatures of recent local adaptations in people from Ibiza Submitted for the degree of Master of Science Diego Alejandro Londoño Correa Supervised by Elena Bosch (Institute of Evolutionary Biology, CSIC-UPF, Barcelona) Carina Schlebusch (internal supervisor at Uppsala University) March 2021 1 Table of Contents ABSTRACT .................................................................................................................................................. 4 INTRODUCTION ......................................................................................................................................... 5 MATERIALS AND METHODS ....................................................................................................................... 8 Data ....................................................................................................................................................... 8 Quality control ....................................................................................................................................... 8 Pruning linked variants, PCA and Admixture ......................................................................................... 9 Selection Tests ....................................................................................................................................... 9 Control for iHS signals in Iberia ........................................................................................................... -

Human Social Genomics in the Multi-Ethnic Study of Atherosclerosis
Getting “Under the Skin”: Human Social Genomics in the Multi-Ethnic Study of Atherosclerosis by Kristen Monét Brown A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Epidemiological Science) in the University of Michigan 2017 Doctoral Committee: Professor Ana V. Diez-Roux, Co-Chair, Drexel University Professor Sharon R. Kardia, Co-Chair Professor Bhramar Mukherjee Assistant Professor Belinda Needham Assistant Professor Jennifer A. Smith © Kristen Monét Brown, 2017 [email protected] ORCID iD: 0000-0002-9955-0568 Dedication I dedicate this dissertation to my grandmother, Gertrude Delores Hampton. Nanny, no one wanted to see me become “Dr. Brown” more than you. I know that you are standing over the bannister of heaven smiling and beaming with pride. I love you more than my words could ever fully express. ii Acknowledgements First, I give honor to God, who is the head of my life. Truly, without Him, none of this would be possible. Countless times throughout this doctoral journey I have relied my favorite scripture, “And we know that all things work together for good, to them that love God, to them who are called according to His purpose (Romans 8:28).” Secondly, I acknowledge my parents, James and Marilyn Brown. From an early age, you two instilled in me the value of education and have been my biggest cheerleaders throughout my entire life. I thank you for your unconditional love, encouragement, sacrifices, and support. I would not be here today without you. I truly thank God that out of the all of the people in the world that He could have chosen to be my parents, that He chose the two of you. -

14 SI D. Chauss Et Al. Table S3 Detected EQ Gene-Specific
Table S3 Detected EQ gene‐specific transcripts statistically decreased in expression during EQ to FP transition. Gene Description log2(Fold Change) p‐value* CC2D2A coiled‐coil and C2 domain containing 2A ‐2.0 1.2E‐03 INSIG2 insulin induced gene 2 ‐2.0 1.2E‐03 ODZ2 teneurin transmembrane protein 2 ‐2.0 1.2E‐03 SEPHS1 selenophosphate synthetase 1 ‐2.0 1.2E‐03 B4GALT6 UDP‐Gal:betaGlcNAc beta 1,4‐ galactosyltransferase, ‐2.0 1.2E‐03 polypeptide 6 CDC42SE2 CDC42 small effector 2 ‐2.0 1.2E‐03 SLIT3 slit homolog 3 (Drosophila) ‐2.1 1.2E‐03 FKBP9 FK506 binding protein 9, 63 kDa ‐2.1 1.2E‐03 ATAD2 ATPase family, AAA domain containing 2 ‐2.1 1.2E‐03 PURH 5‐aminoimidazole‐4‐carboxamide ribonucleotide ‐2.1 1.2E‐03 formyltransferase/IMP cyclohydrolase PLXNA2 plexin A2 ‐2.1 1.2E‐03 CSRNP1 cysteine‐serine‐rich nuclear protein 1 ‐2.1 1.2E‐03 PER2 period circadian clock 2 ‐2.1 1.2E‐03 CERK ceramide kinase ‐2.1 1.2E‐03 NRSN1 neurensin 1 ‐2.1 1.2E‐03 C1H21orf33 ES1 protein homolog, mitochondrial ‐2.1 1.2E‐03 REPS2 RALBP1 associated Eps domain containing 2 ‐2.2 1.2E‐03 TPX2 TPX2, microtubule‐associated, homolog (Xenopus laevis) ‐2.2 1.2E‐03 PPIC peptidylprolyl isomerase C (cyclophilin C) ‐2.2 1.2E‐03 GNG10 guanine nucleotide binding protein (G protein), gamma 10 ‐2.2 1.2E‐03 PHF16 PHD finger protein 16 ‐2.2 1.2E‐03 TMEM108 transmembrane protein 108 ‐2.2 1.2E‐03 MCAM melanoma cell adhesion molecule ‐2.2 1.2E‐03 TLL1 tolloid‐like 1 ‐2.2 1.2E‐03 TMEM194B transmembrane protein 194B ‐2.2 1.2E‐03 PIWIL1 piwi‐like RNA‐mediated gene silencing 1 ‐2.2 1.2E‐03 SORCS1 -

A Point Mutation in HIV-1 Integrase Redirects Proviral Integration Into
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.12.426369; this version posted January 12, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A point mutation in HIV-1 integrase redirects proviral integration into centromeric repeats Shelby Winansa,b,c and Stephen P. Goffa.b,c# aDepartment of Biochemistry and Molecular Biophysics Columbia University Medical Center, New York, NY bDepartment of Microbiology and Immunology Columbia University Medical Center, New York, NY cHoward Hughes Medical Institute, Columbia University, New York, NY #Lead Contact: address correspondence to Stephen P. Goff, [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.01.12.426369; this version posted January 12, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Abstract 2 Retroviruses utilize the viral integrase (IN) protein to integrate a DNA copy of their 3 genome into the host chromosomal DNA. HIV-1 integration sites are highly biased towards 4 actively transcribed genes, likely mediated by binding of the IN protein to specific host 5 factors, particularly LEDGF, located at these gene regions. We here report a dramatic 6 redirection of integration site distribution induced by a single point mutation in HIV-1 IN.