Supplemental Information Figure S1. The
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Anti-ARL4A Antibody (ARG41291)
Product datasheet [email protected] ARG41291 Package: 100 μl anti-ARL4A antibody Store at: -20°C Summary Product Description Rabbit Polyclonal antibody recognizes ARL4A Tested Reactivity Hu, Ms, Rat Tested Application ICC/IF, IHC-P Host Rabbit Clonality Polyclonal Isotype IgG Target Name ARL4A Antigen Species Human Immunogen Recombinant fusion protein corresponding to aa. 121-200 of Human ARL4A (NP_001032241.1). Conjugation Un-conjugated Alternate Names ARL4; ADP-ribosylation factor-like protein 4A Application Instructions Application table Application Dilution ICC/IF 1:50 - 1:200 IHC-P 1:50 - 1:200 Application Note * The dilutions indicate recommended starting dilutions and the optimal dilutions or concentrations should be determined by the scientist. Calculated Mw 23 kDa Properties Form Liquid Purification Affinity purified. Buffer PBS (pH 7.3), 0.02% Sodium azide and 50% Glycerol. Preservative 0.02% Sodium azide Stabilizer 50% Glycerol Storage instruction For continuous use, store undiluted antibody at 2-8°C for up to a week. For long-term storage, aliquot and store at -20°C. Storage in frost free freezers is not recommended. Avoid repeated freeze/thaw cycles. Suggest spin the vial prior to opening. The antibody solution should be gently mixed before use. Note For laboratory research only, not for drug, diagnostic or other use. www.arigobio.com 1/2 Bioinformation Gene Symbol ARL4A Gene Full Name ADP-ribosylation factor-like 4A Background ADP-ribosylation factor-like 4A is a member of the ADP-ribosylation factor family of GTP-binding proteins. ARL4A is similar to ARL4C and ARL4D and each has a nuclear localization signal and an unusually high guaninine nucleotide exchange rate. -

A Database for Human and Yeast Genes Involved in Telomere Maintenance Delia M
Braun et al. BMC Genetics (2018) 19:32 https://doi.org/10.1186/s12863-018-0617-8 DATABASE Open Access TelNet - a database for human and yeast genes involved in telomere maintenance Delia M. Braun, Inn Chung, Nick Kepper, Katharina I. Deeg and Karsten Rippe* Abstract Background: The ends of linear chromosomes, the telomeres, comprise repetitive DNA sequences in complex with proteins that protects them from being processed by the DNA repair machinery. Cancer cells need to counteract the shortening of telomere repeats during replication for their unlimited proliferation by reactivating the reverse transcriptase telomerase or by using the alternative lengthening of telomeres (ALT) pathway. The different telomere maintenance (TM) mechanisms appear to involve hundreds of proteins but their telomere repeat length related activities are only partly understood. Currently, a database that integrates information on TM relevant genes is missing. Description: To provide a resource for studies that dissect TM features, we here introduce the TelNet database at http://www.cancertelsys.org/telnet/. It offers a comprehensive compilation of more than 2000 human and 1100 yeast genes linked to telomere maintenance. These genes were annotated in terms of TM mechanism, associated specific functions and orthologous genes, a TM significance score and information from peer-reviewed literature. This TM information can be retrieved via different search and view modes and evaluated for a set of genes as demonstrated for an exemplary application. Conclusion: TelNet supports the annotation of genes identified from bioinformatics analysis pipelines to reveal possible connections with TM networks. We anticipate that TelNet will be a helpful resource for researchers that study telomeres. -

Temporal Chip-On-Chip of RNA-Polymerase-II to Detect Novel Gene Activation Events During Photoreceptor Maturation
Molecular Vision 2010; 16:252-271 <http://www.molvis.org/molvis/v16/a32> © 2010 Molecular Vision Received 12 July 2009 | Accepted 10 February 2010 | Published 17 February 2010 Temporal ChIP-on-Chip of RNA-Polymerase-II to detect novel gene activation events during photoreceptor maturation Padmaja Tummala, Raghuveer S. Mali, Eduardo Guzman, Xiao Zhang, Kenneth P. Mitton (The first two authors contributed equally to this work.) Eye Research Institute, Oakland University, Rochester, MI Purpose: During retinal development, post-mitotic neural progenitor cells must activate thousands of genes to complete synaptogenesis and terminal maturation. While many of these genes are known, others remain beyond the sensitivity of expression microarray analysis. Some of these elusive gene activation events can be detected by mapping changes in RNA polymerase-II (Pol-II) association around transcription start sites. Methods: High-resolution (35 bp) chromatin immunoprecipitation (ChIP)-on-chip was used to map changes in Pol-II binding surrounding 26,000 gene transcription start sites during photoreceptor maturation of the mouse neural retina, comparing postnatal age 25 (P25) to P2. Coverage was 10–12 kb per transcription start site, including 2.5 kb downstream. Pol-II-active regions were mapped to the mouse genomic DNA sequence by using computational methods (Tiling Analysis Software-TAS program), and the ratio of maximum Pol-II binding (P25/P2) was calculated for each gene. A validation set of 36 genes (3%), representing a full range of Pol-II signal ratios (P25/P2), were examined with quantitative ChIP assays for transcriptionally active Pol-II. Gene expression assays were also performed for 19 genes of the validation set, again on independent samples. -

1 UST College of Science Department of Biological Sciences
UST College of Science Department of Biological Sciences 1 Pharmacogenomics of Myofascial Pain Syndrome An Undergraduate Thesis Submitted to the Department of Biological Sciences College of Science University of Santo Tomas In Partial Fulfillment of the Requirements for the Degree of Bachelor of Science in Biology Jose Marie V. Lazaga Marc Llandro C. Fernandez May 2021 UST College of Science Department of Biological Sciences 2 PANEL APPROVAL SHEET This undergraduate research manuscript entitled: Pharmacogenomics of Myofascial Pain Syndrome prepared and submitted by Jose Marie V. Lazaga and Marc Llandro C. Fernandez, was checked and has complied with the revisions and suggestions requested by panel members after thorough evaluation. This final version of the manuscript is hereby approved and accepted for submission in partial fulfillment of the requirements for the degree of Bachelor of Science in Biology. Noted by: Asst. Prof. Marilyn G. Rimando, PhD Research adviser, Bio/MicroSem 602-603 Approved by: Bio/MicroSem 603 panel member Bio/MicroSem 603 panel member Date: Date: UST College of Science Department of Biological Sciences 3 DECLARATION OF ORIGINALITY We hereby affirm that this submission is our own work and that, to the best of our knowledge and belief, it contains no material previously published or written by another person nor material to which a substantial extent has been accepted for award of any other degree or diploma of a university or other institute of higher learning, except where due acknowledgement is made in the text. We also declare that the intellectual content of this undergraduate research is the product of our work, even though we may have received assistance from others on style, presentation, and language expression. -

Supplemental Information
Supplemental information Dissection of the genomic structure of the miR-183/96/182 gene. Previously, we showed that the miR-183/96/182 cluster is an intergenic miRNA cluster, located in a ~60-kb interval between the genes encoding nuclear respiratory factor-1 (Nrf1) and ubiquitin-conjugating enzyme E2H (Ube2h) on mouse chr6qA3.3 (1). To start to uncover the genomic structure of the miR- 183/96/182 gene, we first studied genomic features around miR-183/96/182 in the UCSC genome browser (http://genome.UCSC.edu/), and identified two CpG islands 3.4-6.5 kb 5’ of pre-miR-183, the most 5’ miRNA of the cluster (Fig. 1A; Fig. S1 and Seq. S1). A cDNA clone, AK044220, located at 3.2-4.6 kb 5’ to pre-miR-183, encompasses the second CpG island (Fig. 1A; Fig. S1). We hypothesized that this cDNA clone was derived from 5’ exon(s) of the primary transcript of the miR-183/96/182 gene, as CpG islands are often associated with promoters (2). Supporting this hypothesis, multiple expressed sequences detected by gene-trap clones, including clone D016D06 (3, 4), were co-localized with the cDNA clone AK044220 (Fig. 1A; Fig. S1). Clone D016D06, deposited by the German GeneTrap Consortium (GGTC) (http://tikus.gsf.de) (3, 4), was derived from insertion of a retroviral construct, rFlpROSAβgeo in 129S2 ES cells (Fig. 1A and C). The rFlpROSAβgeo construct carries a promoterless reporter gene, the β−geo cassette - an in-frame fusion of the β-galactosidase and neomycin resistance (Neor) gene (5), with a splicing acceptor (SA) immediately upstream, and a polyA signal downstream of the β−geo cassette (Fig. -

Functional Annotation of Novel Lineage-Specific Genes Using Co-Expression and Promoter Analysis Charu G Kumar1, Robin E Everts1,3, Juan J Loor1, Harris a Lewin1,2*
Kumar et al. BMC Genomics 2010, 11:161 http://www.biomedcentral.com/1471-2164/11/161 RESEARCH ARTICLE Open Access Functional annotation of novel lineage-specific genes using co-expression and promoter analysis Charu G Kumar1, Robin E Everts1,3, Juan J Loor1, Harris A Lewin1,2* Abstract Background: The diversity of placental architectures within and among mammalian orders is believed to be the result of adaptive evolution. Although, the genetic basis for these differences is unknown, some may arise from rapidly diverging and lineage-specific genes. Previously, we identified 91 novel lineage-specific transcripts (LSTs) from a cow term-placenta cDNA library, which are excellent candidates for adaptive placental functions acquired by the ruminant lineage. The aim of the present study was to infer functions of previously uncharacterized lineage- specific genes (LSGs) using co-expression, promoter, pathway and network analysis. Results: Clusters of co-expressed genes preferentially expressed in liver, placenta and thymus were found using 49 previously uncharacterized LSTs as seeds. Over-represented composite transcription factor binding sites (TFBS) in promoters of clustered LSGs and known genes were then identified computationally. Functions were inferred for nine previously uncharacterized LSGs using co-expression analysis and pathway analysis tools. Our results predict that these LSGs may function in cell signaling, glycerophospholipid/fatty acid metabolism, protein trafficking, regulatory processes in the nucleus, and processes that initiate parturition and immune system development. Conclusions: The placenta is a rich source of lineage-specific genes that function in the adaptive evolution of placental architecture and functions. We have shown that co-expression, promoter, and gene network analyses are useful methods to infer functions of LSGs with heretofore unknown functions. -

Genomic and Transcriptome Analysis Revealing an Oncogenic Functional Module in Meningiomas
Neurosurg Focus 35 (6):E3, 2013 ©AANS, 2013 Genomic and transcriptome analysis revealing an oncogenic functional module in meningiomas XIAO CHANG, PH.D.,1 LINGLING SHI, PH.D.,2 FAN GAO, PH.D.,1 JONATHAN RUssIN, M.D.,3 LIYUN ZENG, PH.D.,1 SHUHAN HE, B.S.,3 THOMAS C. CHEN, M.D.,3 STEVEN L. GIANNOTTA, M.D.,3 DANIEL J. WEISENBERGER, PH.D.,4 GAbrIEL ZADA, M.D.,3 KAI WANG, PH.D.,1,5,6 AND WIllIAM J. MAck, M.D.1,3 1Zilkha Neurogenetic Institute, Keck School of Medicine, University of Southern California, Los Angeles, California; 2GHM Institute of CNS Regeneration, Jinan University, Guangzhou, China; 3Department of Neurosurgery, Keck School of Medicine, University of Southern California, Los Angeles, California; 4USC Epigenome Center, Keck School of Medicine, University of Southern California, Los Angeles, California; 5Department of Psychiatry, Keck School of Medicine, University of Southern California, Los Angeles, California; and 6Division of Bioinformatics, Department of Preventive Medicine, Keck School of Medicine, University of Southern California, Los Angeles, California Object. Meningiomas are among the most common primary adult brain tumors. Although typically benign, roughly 2%–5% display malignant pathological features. The key molecular pathways involved in malignant trans- formation remain to be determined. Methods. Illumina expression microarrays were used to assess gene expression levels, and Illumina single- nucleotide polymorphism arrays were used to identify copy number variants in benign, atypical, and malignant me- ningiomas (19 tumors, including 4 malignant ones). The authors also reanalyzed 2 expression data sets generated on Affymetrix microarrays (n = 68, including 6 malignant ones; n = 56, including 3 malignant ones). -

Nº Ref Uniprot Proteína Péptidos Identificados Por MS/MS 1 P01024
Document downloaded from http://www.elsevier.es, day 26/09/2021. This copy is for personal use. Any transmission of this document by any media or format is strictly prohibited. Nº Ref Uniprot Proteína Péptidos identificados 1 P01024 CO3_HUMAN Complement C3 OS=Homo sapiens GN=C3 PE=1 SV=2 por 162MS/MS 2 P02751 FINC_HUMAN Fibronectin OS=Homo sapiens GN=FN1 PE=1 SV=4 131 3 P01023 A2MG_HUMAN Alpha-2-macroglobulin OS=Homo sapiens GN=A2M PE=1 SV=3 128 4 P0C0L4 CO4A_HUMAN Complement C4-A OS=Homo sapiens GN=C4A PE=1 SV=1 95 5 P04275 VWF_HUMAN von Willebrand factor OS=Homo sapiens GN=VWF PE=1 SV=4 81 6 P02675 FIBB_HUMAN Fibrinogen beta chain OS=Homo sapiens GN=FGB PE=1 SV=2 78 7 P01031 CO5_HUMAN Complement C5 OS=Homo sapiens GN=C5 PE=1 SV=4 66 8 P02768 ALBU_HUMAN Serum albumin OS=Homo sapiens GN=ALB PE=1 SV=2 66 9 P00450 CERU_HUMAN Ceruloplasmin OS=Homo sapiens GN=CP PE=1 SV=1 64 10 P02671 FIBA_HUMAN Fibrinogen alpha chain OS=Homo sapiens GN=FGA PE=1 SV=2 58 11 P08603 CFAH_HUMAN Complement factor H OS=Homo sapiens GN=CFH PE=1 SV=4 56 12 P02787 TRFE_HUMAN Serotransferrin OS=Homo sapiens GN=TF PE=1 SV=3 54 13 P00747 PLMN_HUMAN Plasminogen OS=Homo sapiens GN=PLG PE=1 SV=2 48 14 P02679 FIBG_HUMAN Fibrinogen gamma chain OS=Homo sapiens GN=FGG PE=1 SV=3 47 15 P01871 IGHM_HUMAN Ig mu chain C region OS=Homo sapiens GN=IGHM PE=1 SV=3 41 16 P04003 C4BPA_HUMAN C4b-binding protein alpha chain OS=Homo sapiens GN=C4BPA PE=1 SV=2 37 17 Q9Y6R7 FCGBP_HUMAN IgGFc-binding protein OS=Homo sapiens GN=FCGBP PE=1 SV=3 30 18 O43866 CD5L_HUMAN CD5 antigen-like OS=Homo -

Snp Associations with Tuberculosis Susceptibility in a Ugandan
SNP ASSOCIATIONS WITH TUBERCULOSIS SUSCEPTIBILITY IN A UGANDAN HOUSEHOLD CONTACT STUDY by ALLISON REES BAKER Submitted in partial fulfillment of the requirements For the degree of Master of Science Thesis Advisor: Dr. Catherine M. Stein Department of Epidemiology and Biostatistics CASE WESTERN RESERVE UNIVERSITY August, 2010 CASE WESTERN RESERVE UNIVERSITY SCHOOL OF GRADUATE STUDIES We hereby approve the thesis/dissertation of ______________________________________________________ candidate for the ________________________________degree *. (signed)_______________________________________________ (chair of the committee) ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ ________________________________________________ (date) _______________________ *We also certify that written approval has been obtained for any proprietary material contained therein. Table of Contents Table of Contents...............................................................................................................iii List of Tables ..................................................................................................................... iv Acknowledgements............................................................................................................. v List of Commonly Used Abbreviations ............................................................................. vi Chapter 1: Literature -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

Lin28a/Let-7 Pathway Modulates the Hox Code Via Polycomb Regulation
RESEARCH ARTICLE Lin28a/let-7 pathway modulates the Hox code via Polycomb regulation during axial patterning in vertebrates Tempei Sato1,2,3, Kensuke Kataoka1,3, Yoshiaki Ito1,4, Shigetoshi Yokoyama2,5, Masafumi Inui2,6, Masaki Mori1,7, Satoru Takahashi8, Keiichi Akita9, Shuji Takada2, Hiroe Ueno-Kudoh2,10, Hiroshi Asahara1,2,11,12* 1Department of Systems BioMedicine, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo, Japan; 2Department of Systems BioMedicine, National Research Institute for Child Health and Development, Tokyo, Japan; 3Research Fellow of Japan Society for the Promotion of Science, Tokyo, Japan; 4Research Core, Tokyo Medical and Dental University, Tokyo, Japan; 5Laboratory of Metabolism, National Institutes of Health, Bethesda, United States; 6Laboratory of Animal Regeneration Systemology, Meiji University, Kanagawa, Japan; 7Department of Medical Chemistry, Shiga University of Medical Science, Shiga, Japan; 8Department of Anatomy and Embryology, University of Tsukuba, Ibaraki, Japan; 9Department of Clinical Anatomy, Graduate School of Medical and Dental Sciences, Tokyo Medical and Dental University, Tokyo, Japan; 10Reproduction Center, Yokohama City University, Yokohama, Japan; 11AMED- CREST, Japan Agency for Medical Research and Development (AMED), Tokyo, Japan; 12Department of Molecular Medicine, The Scripps Research Institute, La Jolla, United States Abstract The body plan along the anteroposterior axis and regional identities are specified by *For correspondence: the spatiotemporal expression of Hox genes. Multistep controls are required for their unique [email protected] expression patterns; however, the molecular mechanisms behind the tight control of Hox genes are not fully understood. In this study, we demonstrated that the Lin28a/let-7 pathway is critical for Competing interests: The axial elongation. -
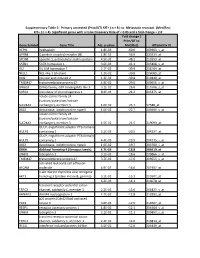
Gene Symbol Gene Title Adj. P-‐Value Fold Change ( Prim/UT to Met
Supplementary Table 2: Primary untreated (Prim/UT) KIT+ ( n = 8 ) vs. Metastatic resistant (Met/Res) KIT+ ( n = 4). Significant genes with a False Discovery Rate of < 0.05 and a fold change > 2.0 Fold change ( Prim/UT to Gene Symbol Gene Title Adj. p-value Met/Res) Affymetrix ID HEPH hephaestin 1.8E-03 -60.6 203903_s_at GPR88 G protein-coupled receptor 88 1.8E-02 -56.0 220313_at SPON1 spondin 1, extracellular matrix protein 4.5E-02 -48.2 213993_at SATB1 SATB homeobox 1 3.6E-03 -41.1 203408_s_at ISL1 ISL LIM homeobox 1 2.7E-02 -39.9 206104_at NELL1 NEL-like 1 (chicken) 1.2E-02 -39.8 206089_at RAI2 retinoic acid induced 2 1.3E-02 -30.8 219440_at TMEM47 transmembrane protein 47 4.9E-02 -29.0 209656_s_at DIRAS3 DIRAS family, GTP-binding RAS-like 3 3.1E-02 -26.8 215506_s_at SCRG1 stimulator of chondrogenesis 1 8.3E-03 -24.2 205475_at solute carrier family 24 (sodium/potassium/calcium SLC24A3 exchanger), member 3 1.6E-02 -23.7 57588_at DIO2 deiodinase, iodothyronine, type II 1.0E-02 -22.7 203699_s_at solute carrier family 24 (sodium/potassium/calcium SLC24A3 exchanger), member 3 1.5E-02 -21.5 219090_at GULP, engulfment adaptor PTB domain GULP1 containing 1 5.2E-03 -20.5 204237_at GULP, engulfment adaptor PTB domain GULP1 containing 1 4.4E-03 -19.9 204235_s_at DIO2 deiodinase, iodothyronine, type II 1.0E-02 -19.7 203700_s_at DKK4 dickkopf homolog 4 (Xenopus laevis) 4.7E-03 -18.8 206619_at LPHN3 latrophilin 3 3.1E-02 -18.6 209866_s_at TMEM47 transmembrane protein 47 3.7E-02 -17.6 209655_s_at activated leukocyte cell adhesion ALCAM molecule 4.9E-02