Dynamic Control of Enhancer Activity Drives Stage-Specific Gene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An in Vivo Screen Identifies PYGO2 As a Driver for Metastatic Prostate Cancer
Published OnlineFirst May 16, 2018; DOI: 10.1158/0008-5472.CAN-17-3564 Cancer Molecular Cell Biology Research An In Vivo Screen Identifies PYGO2 as a Driver for Metastatic Prostate Cancer Xin Lu1,2,3, Xiaolu Pan1, Chang-Jiun Wu4, Di Zhao1, Shan Feng2, Yong Zang5, Rumi Lee1, Sunada Khadka1, Samirkumar B. Amin4, Eun-Jung Jin6, Xiaoying Shang1, Pingna Deng1,Yanting Luo2,William R. Morgenlander2, Jacqueline Weinrich2, Xuemin Lu2, Shan Jiang7, Qing Chang7, Nora M. Navone8, Patricia Troncoso9, Ronald A. DePinho1, and Y. Alan Wang1 Abstract Advanced prostate cancer displays conspicuous chromosomal inhibits prostate cancer cell invasion in vitro and progression of instability and rampant copy number aberrations, yet the identity primary tumor and metastasis in vivo. In clinical samples, PYGO2 of functional drivers resident in many amplicons remain elusive. upregulation associated with higher Gleason score and metastasis Here, we implemented a functional genomics approach to identify to lymph nodes and bone. Silencing PYGO2 expression in patient- new oncogenes involved in prostate cancer progression. Through derived xenograft models impairs tumor progression. Finally, integrated analyses of focal amplicons in large prostate cancer PYGO2 is necessary to enhance the transcriptional activation in genomic and transcriptomic datasets as well as genes upregulated response to ligand-induced Wnt/b-catenin signaling. Together, our in metastasis, 276 putative oncogenes were enlisted into an in vivo results indicate that PYGO2 functions as a driver oncogene in the gain-of-function tumorigenesis screen. Among the top positive 1q21.3 amplicon and may serve as a potential prognostic bio- hits, we conducted an in-depth functional analysis on Pygopus marker and therapeutic target for metastatic prostate cancer. -

GIGANTUS1 (GTS1), a Member of Transducin/WD40 Protein
Gachomo et al. BMC Plant Biology 2014, 14:37 http://www.biomedcentral.com/1471-2229/14/37 RESEARCH ARTICLE Open Access GIGANTUS1 (GTS1), a member of Transducin/WD40 protein superfamily, controls seed germination, growth and biomass accumulation through ribosome-biogenesis protein interactions in Arabidopsis thaliana Emma W Gachomo1,2†, Jose C Jimenez-Lopez3,4†, Lyla Jno Baptiste1 and Simeon O Kotchoni1,2* Abstract Background: WD40 domains have been found in a plethora of eukaryotic proteins, acting as scaffolding molecules assisting proper activity of other proteins, and are involved in multi-cellular processes. They comprise several stretches of 44-60 amino acid residues often terminating with a WD di-peptide. They act as a site of protein-protein interactions or multi-interacting platforms, driving the assembly of protein complexes or as mediators of transient interplay among other proteins. In Arabidopsis, members of WD40 protein superfamily are known as key regulators of plant-specific events, biologically playing important roles in development and also during stress signaling. Results: Using reverse genetic and protein modeling approaches, we characterize GIGANTUS1 (GTS1), a new member of WD40 repeat protein in Arabidopsis thaliana and provide evidence of its role in controlling plant growth development. GTS1 is highly expressed during embryo development and negatively regulates seed germination, biomass yield and growth improvement in plants. Structural modeling analysis suggests that GTS1 folds into a β-propeller with seven pseudo symmetrically arranged blades around a central axis. Molecular docking analysis shows that GTS1 physically interacts with two ribosomal protein partners, a component of ribosome Nop16, and a ribosome-biogenesis factor L19e through β-propeller blade 4 to regulate cell growth development. -

BOP1 (A-18): Sc-87884
SAN TA C RUZ BI OTEC HNOL OG Y, INC . BOP1 (A-18): sc-87884 BACKGROUND CHROMOSOMAL LOCATION Predominantly localized to the nucleolus, BOP1 (Block of proliferation 1 pro - Genetic locus: BOP1 (human) mapping to 8q24.3; Bop1 (mouse) mapping to tein) is a 746 amino acid highly conserved non-ribosomal protein that is in- 15 D3. volved in ribosome biogenesis. Truncation of the amino terminus of BOP1 leads to cell growth arrest in the G 1 phase and specific inhibition of 28S and SOURCE 5.8S rRNA synthesis, as well as a deficit in the cytosolic 60S ribosomal sub - BOP1 (A-18) is an affinity purified rabbit polyclonal antibody raised against a unit. This suggests that BOP1 is involved in the formation of mature rRNAs peptide mapping within an internal region of BOP1 of human origin. and in the biogenesis of the 60S ribosomal subunit. BOP1 physically inter acts with pescadillo (a protein involved in cell proliferation) and enables efficient PRODUCT incorporation of pescadillo into the nucleolar preribosomal complexes, there - by affecting rRNA maturation and the cell cycle. The BOP1-pescadillo com - Each vial contains 100 µg IgG in 1.0 ml of PBS with < 0.1% sodium azide plex is also necessary for biogenesis of 60S ribosomal subunits. Deregulation and 0.1% gelatin. of BOP1 may lead to colorectal tumorigenesis. Blocking peptide available for competition studies, sc-87884 P, (100 µg peptide in 0.5 ml PBS containing < 0.1% sodium azide and 0.2% BSA). REFERENCES 1. Strezoska, Z., Pestov, D.G. and Lau, L.F. 2000. -

The Temporal Mutational and Immune Tumour Microenvironment
www.nature.com/npjbcancer ARTICLE OPEN The temporal mutational and immune tumour microenvironment remodelling of HER2-negative primary breast cancers ✉ Leticia De Mattos-Arruda 1,2,3 , Javier Cortes 4,5,6,7,8, Juan Blanco-Heredia1,2, Daniel G. Tiezzi 3,9, Guillermo Villacampa10, Samuel Gonçalves-Ribeiro 10, Laia Paré11,12,13, Carla Anjos Souza1,2, Vanesa Ortega7, Stephen-John Sammut3,14, Pol Cusco10, Roberta Fasani10, Suet-Feung Chin 3, Jose Perez-Garcia4,5,6, Rodrigo Dienstmann10, Paolo Nuciforo10, Patricia Villagrasa12, Isabel T. Rubio10, Aleix Prat 11,12,13 and Carlos Caldas 3,14 The biology of breast cancer response to neoadjuvant therapy is underrepresented in the literature and provides a window-of- opportunity to explore the genomic and microenvironment modulation of tumours exposed to therapy. Here, we characterised the mutational, gene expression, pathway enrichment and tumour-infiltrating lymphocytes (TILs) dynamics across different timepoints of 35 HER2-negative primary breast cancer patients receiving neoadjuvant eribulin therapy (SOLTI-1007 NEOERIBULIN- NCT01669252). Whole-exome data (N = 88 samples) generated mutational profiles and candidate neoantigens and were analysed along with RNA-Nanostring 545-gene expression (N = 96 samples) and stromal TILs (N = 105 samples). Tumour mutation burden varied across patients at baseline but not across the sampling timepoints for each patient. Mutational signatures were not always conserved across tumours. There was a trend towards higher odds of response and less hazard to relapse when the percentage of 1234567890():,; subclonal mutations was low, suggesting that more homogenous tumours might have better responses to neoadjuvant therapy. Few driver mutations (5.1%) generated putative neoantigens. -

Overexpressed HSF1 Cancer Signature Genes Cluster in Human Chromosome 8Q Christopher Q
Zhang et al. Human Genomics (2017) 11:35 DOI 10.1186/s40246-017-0131-5 PRIMARY RESEARCH Open Access Overexpressed HSF1 cancer signature genes cluster in human chromosome 8q Christopher Q. Zhang1,4, Heinric Williams3,4, Thomas L. Prince3,4† and Eric S. Ho1,2*† Abstract Background: HSF1 (heat shock factor 1) is a transcription factor that is found to facilitate malignant cancer development and proliferation. In cancer cells, HSF1 mediates a set of genes distinct from heat shock that contributes to malignancy. This set of genes is known as the HSF1 Cancer Signature genes or simply HSF1-CanSig genes. HSF1-CanSig genes function and operate differently than typical cancer-causing genes, yet it is involved in fundamental oncogenic processes. Results: By utilizing expression data from 9241 cancer patients, we identified that human chromosome 8q21-24 is a location hotspot for the most frequently overexpressed HSF1-CanSig genes. Intriguingly, the strength of the HSF1 cancer program correlates with the number of overexpressed HSF1-CanSig genes in 8q, illuminating the essential role of HSF1 in mediating gene expression in different cancers. Chromosome 8q21-24 is found under selective pressure in preserving gene order as it exhibits strong synteny among human, mouse, rat, and bovine, although the biological significance remains unknown. Statistical modeling, hierarchical clustering, and gene ontology-based pathway analyses indicate crosstalk between HSF1-mediated responses and pre-mRNA 3′ processing in cancers. Conclusions: Our results confirm the unique role of chromosome 8q mediated by the master regulator HSF1 in cancer cases. Additionally, this study highlights the connection between cellular processes triggered by HSF1 and pre-mRNA 3′ processing in cancers. -
![Floral Induction in Arabidopsis by FLOWERING LOCUS T Requires Direct Repression of BLADE-ON-PETIOLE Genes by the Homeodomain Protein PENNYWISE1[OPEN]](https://docslib.b-cdn.net/cover/2445/floral-induction-in-arabidopsis-by-flowering-locus-t-requires-direct-repression-of-blade-on-petiole-genes-by-the-homeodomain-protein-pennywise1-open-1862445.webp)
Floral Induction in Arabidopsis by FLOWERING LOCUS T Requires Direct Repression of BLADE-ON-PETIOLE Genes by the Homeodomain Protein PENNYWISE1[OPEN]
Floral Induction in Arabidopsis by FLOWERING LOCUS T Requires Direct Repression of BLADE-ON-PETIOLE Genes by the Homeodomain Protein PENNYWISE1[OPEN] Fernando Andrés, Maida Romera-Branchat, Rafael Martínez-Gallegos, Vipul Patel, Korbinian Schneeberger, Seonghoe Jang2, Janine Altmüller, Peter Nürnberg, and George Coupland* Max Planck Institute for Plant Breeding Research, D–50829 Cologne, Germany (F.A., M.R.-B., R.M.-G., V.P., K.S., S.J., G.C.); and Cologne Center for Genomics (J.A., P.N.), Institute of Human Genetics (J.A.), Center for Molecular Medicine Cologne (P.N.), and Cologne Excellence Cluster on Cellular Stress Responses in Aging- Associated Diseases (P.N.), University of Cologne, 50931 Cologne, Germany ORCID IDs: 0000-0003-4736-8876 (F.A.); 0000-0002-6685-5066 (M.R.-B.); 0000-0003-0012-5033 (R.M.-G.); 0000-0001-5018-3480 (S.J.); 0000-0001-6988-4172 (G.C.). Flowers form on the flanks of the shoot apical meristem (SAM) in response to environmental and endogenous cues. In Arabidopsis (Arabidopsis thaliana), the photoperiodic pathway acts through FLOWERING LOCUS T (FT) to promote floral induction in response to day length. A complex between FT and the basic leucine-zipper transcription factor FD is proposed to form in the SAM, leading to activation of APETALA1 and LEAFY and thereby promoting floral meristem identity. We identified mutations that suppress FT function and recovered a new allele of the homeodomain transcription factor PENNYWISE (PNY). Genetic and molecular analyses showed that ectopic expression of BLADE-ON-PETIOLE1 (BOP1) and BOP2, which encode transcriptional coactivators, in the SAM during vegetative development, confers the late flowering of pny mutants. -

PDF of Eric Lecuyer's Talk
Gene Expression Regulation in Subcellular Space Eric Lécuyer, PhD Associate Professor and Axis Director, RNA Biology Lab Systems Biology Axis, IRCM Associate Research Professor, Département de Biochimie Université de Montréal Associate Member, Division of Experimental Medicine, McGill University VizBi 2018 Meeting The Central Dogma in Subcellular Space Crick, Nature 227: 561 (1970) Biological Functions of Localized mRNAs mRNA Protein Extracellular Vesicles Cody et al. (2013). WIREs Dev Biol Raposo and Stoorvogel (2013) J Cell Biol Cis-Regulatory RNA Localization Elements Van De Bor and Davis, 2004. Curr.Opin.Cell.Biol. Global Screen for Localized mRNAs in Drosophila http://fly-fish.ccbr.utoronto.ca (Lécuyer et al. Cell, 2007) Diverse RNA Subcellular Localization Patterns RNA DNA Correlations in mRNA-Protein Localization Localization Patterns Terms Ontology Gene RNA Protein DNA (Lécuyer et al. Cell, 2007) Models and Approaches to Decipher the mRNA Localization Pathways Drosophila & RNA/Protein High-Content Screening Human Cell Models Imaging & RNA Sequencing + + Cell Fractionation and RNA Sequencing (CeFra-seq) to Study Global RNA Distribution Cell Fractionation-Seq to Study RNA Localization RNA and Protein Extraction, RiboDepletion or PolyA+ RNA-seq and MS profiling (K562, HepG2 and D17) Wang et al (2012) Cell https://www.encodeproject.org/ Lefebvre et al (2017) Methods Benoît Bouvrette et al (2018) RNA Interesting Examples of RNA Localization ANKRD52 (mRNA and ciRNA) Total Nuclear Cytosolic ciRNA Membrane Insoluble mRNA ANKRD52 DANCR -

Renoprotective Effect of Combined Inhibition of Angiotensin-Converting Enzyme and Histone Deacetylase
BASIC RESEARCH www.jasn.org Renoprotective Effect of Combined Inhibition of Angiotensin-Converting Enzyme and Histone Deacetylase † ‡ Yifei Zhong,* Edward Y. Chen, § Ruijie Liu,*¶ Peter Y. Chuang,* Sandeep K. Mallipattu,* ‡ ‡ † | ‡ Christopher M. Tan, § Neil R. Clark, § Yueyi Deng, Paul E. Klotman, Avi Ma’ayan, § and ‡ John Cijiang He* ¶ *Department of Medicine, Mount Sinai School of Medicine, New York, New York; †Department of Nephrology, Longhua Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai, China; ‡Department of Pharmacology and Systems Therapeutics and §Systems Biology Center New York, Mount Sinai School of Medicine, New York, New York; |Baylor College of Medicine, Houston, Texas; and ¶Renal Section, James J. Peters Veterans Affairs Medical Center, New York, New York ABSTRACT The Connectivity Map database contains microarray signatures of gene expression derived from approximately 6000 experiments that examined the effects of approximately 1300 single drugs on several human cancer cell lines. We used these data to prioritize pairs of drugs expected to reverse the changes in gene expression observed in the kidneys of a mouse model of HIV-associated nephropathy (Tg26 mice). We predicted that the combination of an angiotensin-converting enzyme (ACE) inhibitor and a histone deacetylase inhibitor would maximally reverse the disease-associated expression of genes in the kidneys of these mice. Testing the combination of these inhibitors in Tg26 mice revealed an additive renoprotective effect, as suggested by reduction of proteinuria, improvement of renal function, and attenuation of kidney injury. Furthermore, we observed the predicted treatment-associated changes in the expression of selected genes and pathway components. In summary, these data suggest that the combination of an ACE inhibitor and a histone deacetylase inhibitor could have therapeutic potential for various kidney diseases. -

Chen B, Dragomir MP, Fabr
This is the peer reviewed accepted manuscript of: Chen B, Dragomir MP, Fabris L, Bayraktar R, Knutsen E, Liu X, Tang C, Li Y, Shimura T, Ivkovic TC, De los Santos MC, Anfossi S, Shimizu M, Shah MY, Ling H, Shen P, Multani AS, Pardini B, Burks JK, Katayama H, Reineke LC, Huo L, Syed M, Song S, Ferracin M, Oki E, Fromm B, Ivan C, Bhuvaneshwar K, Gusev Y, Mimori K, Menter D, Sen S, Matsuyama T, Uetake H, Vasilescu C, Kopetz S, Parker-Thornburg J, Taguchi A, Hanash SM, Girnita L, Slaby O, Goel A, Varani G, Gagea M, Li C, Ajani JA, Calin GA, The Long Noncoding RNA CCAT2 induces chromosomal instability through BOP1 - AURKB signaling, Gastroenterology (2020), Final version available at: https://doi.org/10.1053/j.gastro.2020.08.01 Rights / License: The terms and conditions for the reuse of this version of the manuscript are specified in the publishing policy. For all terms of use and more information see the publisher's website. This item was downloaded from IRIS Università di Bologna (https://cris.unibo.it/) When citing, please refer to the published version. The Long Noncoding RNA CCAT2 induces chromosomal instability through BOP1 - AURKB signaling Baoqing Chen1 ,2 ,29 *, Mihnea P. Dragomir1 ,3 *, Linda Fabris1 *, Recep Bayraktar1 *, Erik Knutsen1, 4, Xu Liu1, 5, Changyan Tang6, Yongfeng Li1, Tadanobu Shimura7, Tina Catela Ivkovic8, Mireia Cruz De los Santos1, Simone Anfossi1, Masayoshi Shimizu1, Maitri Y. Shah1, Hui Ling1, Peng Shen1 ,9, Asha S. Multani10, Barbara Pardini1, 30, 31, Jared K. Burks11, Hiroyuki Katayama12, Lucas C. -

The BLADE-ON-PETIOLE 1 Gene Controls Leaf Pattern Formation Through the Modulation of Meristematic Activity in Arabidopsis
Development 130, 161-172 161 © 2003 The Company of Biologists Ltd doi:10.1242/dev.00196 The BLADE-ON-PETIOLE 1 gene controls leaf pattern formation through the modulation of meristematic activity in Arabidopsis Chan Man Ha1, Gyung-Tae Kim2,*, Byung Chul Kim1, Ji Hyung Jun1, Moon Soo Soh1,†, Yoshihisa Ueno3, Yasunori Machida3, Hirokazu Tsukaya2 and Hong Gil Nam1,‡ 1Division of Molecular Life Science, Pohang University of Science and Technology, San 31, Hyoja-dong, Pohang, Kyungbuk, 790- 784, Korea 2National Institute for Basic Biology/Center for Integrative Bioscience, Myodaiji-cho, Okazaki 444-8585, Japan 3Division of Biological Science, Graduate School of Science, Nagoya University, Chikusa-ku, Nagoya 464-8602, Japan *Present address: Department of Life Science and Resources, Dong-A University, Hadan-2-dong 840, Busan, 604-714, Korea †Present address: Kumho Life and Environmental Science Laboratory, Oryong-dong, Gwangju, 500-712, Korea ‡Author for correspondence (e-mail: [email protected]) Accepted 8 October 2002 SUMMARY The plant leaf provides an ideal system to study the within the context of the leaf proximodistality, mechanisms of organ formation and morphogenesis. The dorsoventrality and heteroblasty. BOP1 appears to regulate key factors that control leaf morphogenesis include the meristematic activity in organs other than leaves, since the timing, location and extent of meristematic activity during mutation also causes some ectopic outgrowths on stem cell division and differentiation. We identified an surfaces and at the base of floral organs. Three class I knox Arabidopsis mutant in which the regulation of meristematic genes, i.e., KNAT1, KNAT2 and KNAT6, were expressed activities in leaves was aberrant. -

STRUCTURAL and FUNCTIONAL STUDY of the INTERACTION BETWEEN Ki67 FHA DOMAIN and NIFK
STRUCTURAL AND FUNCTIONAL STUDY OF THE INTERACTION BETWEEN Ki67 FHA DOMAIN AND NIFK DISSERTATION Presented in Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By Haiyan Song, M. S. ***** The Ohio State University 2007 Dissertation Committee: Approved by Professor Ming-Daw Tsai, Advisor Professor Dale D. Vandre, Co-advisor _________________________________ Professor Ross E. Dalbey Advisor Ohio State Biochemistry Graduate Program Professor Sissy M. Jhiang ABSTRACT Both structural biology and cell biology approaches have been used to study the interaction between the FHA domain from Ki67 and a nucleolar protein NIFK. Ki67 is a nuclear protein that is over-expressed in proliferating cells (G 1, S, G 2 and M- phase), but is extremely down-regulated in quiescent cells (G 0). It is believed to be involved in the regulation of rRNA gene transcription and cell proliferation. However, little is known about the precise function of the protein, due to its large size and lack of homology to other proteins with known functions. In this dissertation, efforts have been made to understand both the structural basis and the biological function(s) of the interaction between the FHA domain from the N-terminal of Ki67 and the nucleolar protein NIFK. Previous studies showed that Ki67 FHA domain binds to a phosphopeptide consisting of NIFK amino acid residues 226 to 269. Preliminary results from protein kinase assays also showed that NIFK (226-269) can be sequentially phosphorylated by kinases CDK1/cyclin B and GSK3 β at Thr238 and Thr234, respectively, in vitro . -
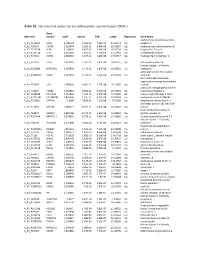
Table S1. the Statistical Metrics for Key Differentially Expressed Genes (Degs)
Table S1. The statistical metrics for key differentially expressed genes (DEGs) Gene Agilent Id Symbol logFC pValue FDR tvalue Regulation Gene Name oxidized low density lipoprotein A_24_P124624 OLR1 2.458429 1.19E-13 7.25E-10 24.04241 Up receptor 1 A_23_P90273 CHST8 2.622464 3.85E-12 6.96E-09 19.05867 Up carbohydrate sulfotransferase 8 A_23_P217528 KLF8 2.109007 4.85E-12 7.64E-09 18.76234 Up Kruppel like factor 8 A_23_P114740 CFH 2.651636 1.85E-11 1.79E-08 17.13652 Up complement factor H A_23_P34031 XAGE2 2.000935 2.04E-11 1.81E-08 17.02457 Up X antigen family member 2 A_23_P27332 TCF4 1.613097 2.32E-11 1.87E-08 16.87275 Up transcription factor 4 histone cluster 1 H1 family A_23_P250385 HIST1H1B 2.298658 2.47E-11 1.87E-08 16.80362 Up member b abnormal spindle microtubule A_33_P3288159 ASPM 2.162032 2.79E-11 2.01E-08 16.66292 Up assembly H19, imprinted maternally expressed transcript (non-protein A_24_P52697 H19 1.499364 4.09E-11 2.76E-08 16.23387 Up coding) potassium voltage-gated channel A_24_P31627 KCNB1 2.289689 6.65E-11 3.97E-08 15.70253 Up subfamily B member 1 A_23_P214168 COL12A1 2.155835 7.59E-11 4.15E-08 15.56005 Up collagen type XII alpha 1 chain A_33_P3271341 LOC388282 2.859496 7.61E-11 4.15E-08 15.55704 Up uncharacterized LOC388282 A_32_P150891 DIAPH3 2.2068 7.83E-11 4.22E-08 15.5268 Up diaphanous related formin 3 zinc finger protein 185 with LIM A_23_P11025 ZNF185 1.385721 8.74E-11 4.59E-08 15.41041 Up domain heat shock protein family B A_23_P96872 HSPB11 1.887166 8.94E-11 4.64E-08 15.38599 Up (small) member 11 A_23_P107454