Dificlir, INN-Fidaxomicin
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Management of Adult Clostridium Difficile Digestive Contaminations: a Literature Review
European Journal of Clinical Microbiology & Infectious Diseases (2019) 38:209–231 https://doi.org/10.1007/s10096-018-3419-z REVIEW Management of adult Clostridium difficile digestive contaminations: a literature review Fanny Mathias1 & Christophe Curti1,2 & Marc Montana1,3 & Charléric Bornet4 & Patrice Vanelle 1,2 Received: 5 October 2018 /Accepted: 30 October 2018 /Published online: 29 November 2018 # Springer-Verlag GmbH Germany, part of Springer Nature 2018 Abstract Clostridium difficile infections (CDI) dramatically increased during the last decade and cause a major public health problem. Current treatments are limited by the high disease recurrence rate, severity of clinical forms, disruption of the gut microbiota, and colonization by vancomycin-resistant enterococci (VRE). In this review, we resumed current treatment options from official recommendation to promising alternatives available in the management of adult CDI, with regard to severity and recurring or non-recurring character of the infection. Vancomycin remains the first-line antibiotic in the management of mild to severe CDI. The use of metronidazole is discussed following the latest US recommendations that replaced it by fidaxomicin as first-line treatment of an initial episode of non-severe CDI. Fidaxomicin, the most recent antibiotic approved for CDI in adults, has several advantages compared to vancomycin and metronidazole, but its efficacy seems limited in cases of multiple recurrences. Innovative therapies such as fecal microbiota transplantation (FMT) and antitoxin antibodies were developed to limit the occurrence of recurrence of CDI. Research is therefore very active, and new antibiotics are being studied as surotomycin, cadazolid, and rinidazole. Keywords Clostridiumdifficile .Fidaxomicin .Fecalmicrobiotatransplantation .Antitoxinantibodies .Surotomycin .Cadazolid Introduction (Fig. -

Prediction of Premature Termination Codon Suppressing Compounds for Treatment of Duchenne Muscular Dystrophy Using Machine Learning
Prediction of Premature Termination Codon Suppressing Compounds for Treatment of Duchenne Muscular Dystrophy using Machine Learning Kate Wang et al. Supplemental Table S1. Drugs selected by Pharmacophore-based, ML-based and DL- based search in the FDA-approved drugs database Pharmacophore WEKA TF 1-Palmitoyl-2-oleoyl-sn-glycero-3- 5-O-phosphono-alpha-D- (phospho-rac-(1-glycerol)) ribofuranosyl diphosphate Acarbose Amikacin Acetylcarnitine Acetarsol Arbutamine Acetylcholine Adenosine Aldehydo-N-Acetyl-D- Benserazide Acyclovir Glucosamine Bisoprolol Adefovir dipivoxil Alendronic acid Brivudine Alfentanil Alginic acid Cefamandole Alitretinoin alpha-Arbutin Cefdinir Azithromycin Amikacin Cefixime Balsalazide Amiloride Cefonicid Bethanechol Arbutin Ceforanide Bicalutamide Ascorbic acid calcium salt Cefotetan Calcium glubionate Auranofin Ceftibuten Cangrelor Azacitidine Ceftolozane Capecitabine Benserazide Cerivastatin Carbamoylcholine Besifloxacin Chlortetracycline Carisoprodol beta-L-fructofuranose Cilastatin Chlorobutanol Bictegravir Citicoline Cidofovir Bismuth subgallate Cladribine Clodronic acid Bleomycin Clarithromycin Colistimethate Bortezomib Clindamycin Cyclandelate Bromotheophylline Clofarabine Dexpanthenol Calcium threonate Cromoglicic acid Edoxudine Capecitabine Demeclocycline Elbasvir Capreomycin Diaminopropanol tetraacetic acid Erdosteine Carbidopa Diazolidinylurea Ethchlorvynol Carbocisteine Dibekacin Ethinamate Carboplatin Dinoprostone Famotidine Cefotetan Dipyridamole Fidaxomicin Chlormerodrin Doripenem Flavin adenine dinucleotide -

Antibiotic Fidaxomicin Is an Rdrp Inhibitor As a Potential New Therapeutic Agent Against Zika Virus
Yuan et al. BMC Medicine (2020) 18:204 https://doi.org/10.1186/s12916-020-01663-1 RESEARCH ARTICLE Open Access Antibiotic fidaxomicin is an RdRp inhibitor as a potential new therapeutic agent against Zika virus Jie Yuan1,2,3†, Jianchen Yu2,3,4†, Yun Huang3, Zhenjian He2,5, Jia Luo6, Yun Wu2,4,7, Yingchun Zheng8, Jueheng Wu2,4,7, Xun Zhu2,4, Haihe Wang1 and Mengfeng Li2,3,4* Abstract Background: Zika virus (ZIKV) infection is a global health problem, and its complications, including congenital Zika syndrome and Guillain-Barré syndrome, constitute a continued threat to humans. Unfortunately, effective therapeutics against ZIKV infection are not available thus far. Methods: We screened the compounds collection consisting of 1789 FDA-approved drugs by a computational docking method to obtain anti-ZIKV candidate compounds targeting ZIKV RNA-dependent RNA polymerase (RdRp). SPR (BIAcore) assay was employed to demonstrate the candidate compounds’ direct binding to ZIKV RdRp, and polymerase activity assay was used to determine the inhibitory effect on ZIKV RdRp-catalyzed RNA synthesis. The antiviral effects on ZIKV in vitro and in vivo were detected in infected cultured cells and in Ifnar1−/− mice infected by ZIKV virus using plaque assay, western blotting, tissue immunofluorescence, and immunohistochemistry. Results: Here, we report that a first-in-class macrocyclic antibiotic, which has been clinically used to treat Clostridium difficile infection, fidaxomicin, potently inhibits ZIKV replication in vitro and in vivo. Our data showed that fidaxomicin was effective against African and Asian lineage ZIKV in a wide variety of cell lines of various tissue origins, and prominently suppressed ZIKV infection and significantly improved survival of infected mice. -

IEHP Medi-Cal Prior Authorization Criteria February 2020
IEHP Medi-Cal Prior Authorization Criteria February 2020 Generic Brand Criteria Covered Uses: Osteoporosis Exclusion Criteria: N/A Required Medical Information: Must meet all of the following requirements: a. Documentation of a T-score less than -2.5 at the lumbar spine, hip (total hip or femoral neck), or radius (one-third radius site). b. Documented inadequate response (e.g. greater than 3 percent decrease in bone mineral density from baseline, fracture from minimal trauma)while receiving the following, or clinically significant adverse effects to all of the following: abaloparatide Tymlos i. An oral bisphosphonate (e.g. alendronate) ii. An intravenous bisphosphonate (e.g. zoledronic acid) iii. Prolia c. Patient is concurrently receiving calcium and vitamin D supplement. d. The combined duration of treatment with any parathyroid hormone analogs has not exceeded a lifetime maximum of 24 months (i.e. abaloparatide and teriparatide) Age Restrictions: N/A Prescriber Restrictions: N/A abobotulinum toxin Dysport Please refer to Botulinum Toxin Drug Class Prior Authorization Criteria A Covered Uses: Herpes labialis or herpes febrilis (cold sore) Exclusion Criteria: N/A acyclovir 5% topical Required Medical Information: Must meet the following requirement: cream a. Failure or clinically significant adverse effects to the alternative: Abreva Age Restrictions: Must be age 12 or older Prescriber Restrictions: N/A Detailed Prior Authorization criteria can be found at: https://www.iehp.org/en/providers/pharmacy-services/rx-pa-drug- treatment-criteria Generic Brand Criteria Covered Uses: Must meet "1" of the following: a. Genital herpes simplex virus infection (HSV) b. Non-life threatening mucocutaneous herpes simplex virus infection, patient immunocompromised acyclovir topical Exclusion Criteria: N/A Required Medical Information: Must meet the following requirement: ointment a. -

DIFICID® (Fidaxomicin) Oral
PHARMACY COVERAGE GUIDELINES ORIGINAL EFFECTIVE DATE: 7/16/2015 SECTION: DRUGS LAST REVIEW DATE: 8/19/2021 LAST CRITERIA REVISION DATE: 8/19/2021 ARCHIVE DATE: DIFICID® (fidaxomicin) oral Coverage for services, procedures, medical devices and drugs are dependent upon benefit eligibility as outlined in the member's specific benefit plan. This Pharmacy Coverage Guideline must be read in its entirety to determine coverage eligibility, if any. This Pharmacy Coverage Guideline provides information related to coverage determinations only and does not imply that a service or treatment is clinically appropriate or inappropriate. The provider and the member are responsible for all decisions regarding the appropriateness of care. Providers should provide BCBSAZ complete medical rationale when requesting any exceptions to these guidelines. The section identified as “Description” defines or describes a service, procedure, medical device or drug and is in no way intended as a statement of medical necessity and/or coverage. The section identified as “Criteria” defines criteria to determine whether a service, procedure, medical device or drug is considered medically necessary or experimental or investigational. State or federal mandates, e.g., FEP program, may dictate that any drug, device or biological product approved by the U.S. Food and Drug Administration (FDA) may not be considered experimental or investigational and thus the drug, device or biological product may be assessed only on the basis of medical necessity. Pharmacy Coverage Guidelines are subject to change as new information becomes available. For purposes of this Pharmacy Coverage Guideline, the terms "experimental" and "investigational" are considered to be interchangeable. BLUE CROSS®, BLUE SHIELD® and the Cross and Shield Symbols are registered service marks of the Blue Cross and Blue Shield Association, an association of independent Blue Cross and Blue Shield Plans. -

Fidaxomicin Related Metabolic Acidosis
ABSTRACT NUMBER FIDAXOMICIN RELATED METABOLIC 5PSQ-121 ACIDOSIS: A CASE REPORT B. Serna Serrano1, A. Serrano Martínez 2, A. Valladolid Walsh 1, I. Pérez Alpuente 1, V. Lerma Gaude 1, M. Clemente Andújar 1, R. Aldaz Francés 1 Section 5: Patient Safety 1Complejo Hospitalario Universitario de Albacete, Pharmacy, Albacete, Spain. and Quality Assurance 2Complejo Hospitalario Universitario de Albacete, Hematology, Albacete, Spain. ATC code: A07 - Antidiarrheals, intestinal antiinflammatory/antiinfective agents BACKGROUND AND IMPORTANCE Fidaxomicin is a macrolide antibiotic used to treat intestinal Clostridium difficile (CD) infection in case of absence to metronidazole or vancomycin treatments. On the other hand, pharmacovigilance collects information, analyzes and notifies cases of suspected Adverse Drug Reactions (ADRs) in order to prevent them in the future. AIM AND To describe a case of metabolic acidosis OBJECTIVES in a patient treated with fidaxomicin and establish it is possible association. MATERIALS AND METHODS We describe the case of an 82-year-old male, diagnosed with multiple myeloma and treated with two full cycles of bortezomib-dexamethasone. He was referred to the Emergency department after presenting melenic diarrhea for one week. As a result, he was hospitalized and diagnosed with upper gastrointestinal bleeding, acute prerenal renal failure, mild thrombopenia, hypokalemia and hyponatremia. After fluid and electrolyte stabilization, it was decided to start with fidaxomicin 200 mg/12h due to fever, confusional syndrome, persistence of diarrhea and positive CD toxin test. The following constants were measured to confirm metabolic - acidosis: gas level of bicarbonate (HCO3 ), partial pressure of carbon dioxide (pCO2), hydrogen ion potential (pH) and anion GAP. The degree of drug/adverse reaction causality was evaluated using the Naranjo algorithm. -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

Telephone Request Service EAP Criteria
Telephone Request Service Reimbursement Criteria Exceptional Access Program, Ministry of Health and Long-Term Care Disclaimer: The information in this document is updated on a regular basis. Although we strive to ensure that all information is accurate at the time of posting, please be aware that some items may be subject to change from time-to-time. The information provided in this document and website is intended for information purposes only and does not provide any medical diagnosis, symptom assessment, health counseling or medical opinion for individual users. This information also does not constitute medical advice for physicians or patients. For more detailed information on prescription drugs, please consult a qualified healthcare professional. Last updated August, 2015. The EAP response letter will list the specific drug, strength and dosage form that will be approved. Refer to the formulary for a list of interchangeable drug products that may be dispensed. 2 Table of contents TELEPHONE REQUEST SERVICE REIMBURSEMENT CRITERIA ............................. 1 Exceptional Access Program, Ministry of Health and Long-Term Care ................... 1 TABLE OF CONTENTS .................................................................................................. 3 INTRODUCTION ............................................................................................................. 6 ANTIBIOTICS ................................................................................................................. 7 Cefazolin ....................................................................................................................... -
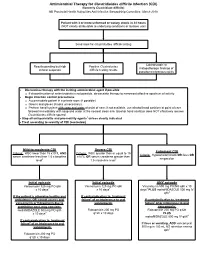
Clostridium Difficile Infection
Antimicrobial Therapy for Clostridioides difficile Infection (CDI) (formerly Clostridium difficile) NB Provincial Health Authorities Anti-Infective Stewardship Committee, March 2019 Patient with 3 or more unformed or watery stools in 24 hours (NOT clearly attributable to underlying conditions or laxative use) Send stool for Clostridioides difficile testing Colonoscopic or Results pending but high Positive Clostridioides histopathologic findings of clinical suspicion difficile testing results pseudomembranous colitis Discontinue therapy with the inciting antimicrobial agent if possible o If discontinuation of antimicrobials is not possible, de-escalate therapy to narrowest effective spectrum of activity Begin infection control precautions o Accommodate patient in a private room (if possible) o Gowns and gloves (masks unnecessary) o Perform hand hygiene with soap and water at point of care; if not available, use alcohol hand sanitizer at point of care followed immediately with soap and water at the nearest clean sink (alcohol hand sanitizer does NOT effectively remove Clostridioides difficile spores) Stop all anti-peristaltic and pro-motility agents1 unless clearly indicated Treat according to severity of CDI (see below) Mild-to-moderate CDI Severe CDI Fulminant CDI Criteria: WBC lower than 15 x109/L AND Criteria: WBC greater than or equal to 15 Criteria: Hypotension/shock OR ileus OR serum creatinine less than 1.5 x baseline x109/L OR serum creatinine greater than megacolon level2 1.5 x baseline level2 Initial episode Initial episode -

Absorption Pharmacokinetics
Conflicts of Interest Top 10 Prescribed Medications . Pharmaceutical Company Speakers Bureau: in Pediatrics None . Pharmaceutical Consulting: None . Medical Device or Lab Consulting: None Anthony J. Busti, MD, PharmD, BSN, FNLA, FAHA . Stock or Financial Interests in Pharmaceutical Faculty, High‐Yield MED Reviews and Medical Device Companies: None Editor‐in‐Chief, Pharmacology Weekly San Antonio, TX . Drug Company Funded Research: None Emergency Medicine Residency Program Johns Hopkins Hospital Baltimore, MD 2 Pediatrics: Absorption . Oral: The Pediatric Patient – Gastric fluids are essentially neutral in the first hrs of life; by the 2nd yr of life gastric output is equal to adult Pharmacokinetics & Pharmacodynamics on per kg basis. – pH‐dependent passive diffusion • full‐term infant: declines from 6‐8 at birth, 1‐3 after 24 hours – Gastric‐emptying time • Not significantly slower, though may not reach adult times until 6‐8 months. 3 4 Pharmacokinetics Pharmacokinetics: Clearance Pathways . First Pass Metabolism: NAT – Clinical Application: Esterase FMO • Lidocaine vs. mexiletine (Mexitil) MAO • Propranolol (Inderal) • Meperidine (Demerol) UGT ≈ 35% CYP450 – Influenced by: • Influx & efflux transporters • Phase I (CYP450) mediated metabolism • Phase II mediated metabolism FMO = flavin monooxygenase MAO = monoamine oxidase NAT = n-acetyltransferase UGT = Uridine glucuronosyltransferase 6 5 Williams J et al. Drug Metab Disp 2004;32:1201-08. NOTE: NOT ALL drugs require transporters to get across cell membranes! Some will make it across by diffusion depending on the charge and degree NOTE: NOT ALL drugs require transporters to get across cell membranes! Some will make it across by diffusion depending on of lipophilicity. 7 the charge and degree of lipophilicity. 8 Pediatrics: Distribution Pharmacokinetics . -

OUH Formulary Approved for Use in Breast Surgery
Oxford University Hospitals NHS Foundation Trust Formulary FORMULARY (Y): the medicine can be used as per its licence. RESTRICTED FORMULARY (R): the medicine can be used as per the agreed restriction. NON-FORMULARY (NF): the medicine is not on the formulary and should not be used unless exceptional approval has been obtained from MMTC. UNLICENSED MEDICINE – RESTRICTED FORMULARY (UNR): the medicine is unlicensed and can be used as per the agreed restriction. SPECIAL MEDICINE – RESTRICTED FORMULARY (SR): the medicine is a “special” (unlicensed) and can be used as per the agreed restriction. EXTEMPORANEOUS PREPARATION – RESTRICTED FORMULARY (EXTR): the extemporaneous preparation (unlicensed) can be prepared and used as per the agreed restriction. UNLICENSED MEDICINE – NON-FORMULARY (UNNF): the medicine is unlicensed and is not on the formulary. It should not be used unless exceptional approval has been obtained from MMTC. SPECIAL MEDICINE – NON-FORMULARY (SNF): the medicine is a “special” (unlicensed) and is not on the formulary. It should not be used unless exceptional approval has been obtained from MMTC. EXTEMPORANEOUS PREPARATION – NON-FORMULARY (EXTNF): the extemporaneous preparation (unlicensed) cannot be prepared and used unless exceptional approval has been obtained from MMTC. CLINICAL TRIALS (C): the medicine is clinical trial material and is not for clinical use. NICE TECHNOLOGY APPRAISAL (NICETA): the medicine has received a positive appraisal from NICE. It will be available on the formulary from the day the Technology Appraisal is published. Prescribers who wish to treat patients who meet NICE criteria, will have access to these medicines from this date. However, these medicines will not be part of routine practice until a NICE TA Implementation Plan has been presented and approved by MMTC (when the drug will be given a Restricted formulary status). -

Drug-Induced Ototoxicity
Continuing Education Drug-Induced Ototoxicity Authors: Erin Bilgili Pharm.D. Harrison School of Pharmacy, Auburn University Jarrid Casimir Pharm.D. Harrison School of Pharmacy, Auburn University Kelli Pickard Pharm.D. Harrison School of Pharmacy, Auburn University Corresponding Author: Wesley Lindsey, Pharm.D. Associate Clinical Professor of Pharmacy Practice Drug Information and Learning Resource Center Harrison School of Pharmacy, Auburn University Universal Activity #: 0178-0000-17-101-H01-P | 1.25 contact hours (.125 CEUs) Initial Release Date: August 7, 2017 | Expires: May 7, 2020 Learning Objectives: After this article, the reader should be able to... • Define ototoxicity and describe the risk factors • Identify the most common classes of ototoxic medications • Discuss the different mechanisms of ototoxicity • Develop a patient-specific monitoring plan • Refer a patient to additional resources if needed Alabama Pharmacy Association | 334.271.4222 | www.aparx.org | [email protected] 1 What is ototoxicity? middle ear, and inner ear. Figure 1 illustrates the Ototoxicity is defined by Hawkins as “the tendency anatomy of the ear and its associated structures. of certain therapeutic agents and other chemical ● Outer ear: external portion of the ear, substances to cause functional impairment and consisting of the pinna, or auricle, and the ear cellular degeneration of the tissues of the inner ear, canal. and especially of the end organs and neurons of the ● Middle ear: includes the eardrum and three cochlear and vestibular divisions of the eighth cranial tiny bones of the middle ear, ending at the nerve”.1 This functional impairment and cellular round window that leads to the inner ear. degeneration can lead to ringing in the ear (tinnitus), ● Inner ear: contains both the organ of hearing hearing loss, or balance disorders.2 Any drug with (cochlea) and the organ of balance the potential to cause toxic effects to the structures of (vestibulum).