Chapter 176: Sensorineural Hearing Loss in Children
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Assessment of Bone Conduction Thresholds After Surgical Treatment in Patients with Labyrinthine Fistula
Turkish Archives of Otorhinolaryngology Turk Arch Otorhinolaryngol 2018; 56(2): 89-94 89 Türk Otorinolarengoloji Arşivi Assessment of Bone Conduction Thresholds After Surgical Treatment in Patients with Labyrinthine Fistula Müzeyyen Yıldırım Baylan1 , Ümit Yılmaz1 , Zeki Akkuş2 , İsmail Topçu1 1Department of Otorhinolaryngology, Dicle University School of Medicine, Diyarbakır, Turkey Original Investigation 2Department of Biostatistics, Dicle University School of Medicine, Diyarbakır, Turkey Abstract Objective: This study aimed to analyze the bone con- years. In the post-operative period, it was possib- duction thresholds before and after surgery in chronic le to conduct audiological follow-up on 20 patients. otitis media patients with cholesteatoma who had In these follow-ups, 16 patients showed no change labyrinthine fistula and whose cholesteatoma matrix in bone conduction thresholds, two patients showed had been completely cleaned. worsening, and two showed improvement. When Methods: The study was performed between 2013 pre- and post-operative bone conduction thresholds to 2017 with 23 chronic otitis media patients who at each frequency were compared separately, no sig- had labyrinthine fistula with cholesteatoma and who were operated at the Department of Otorhinolar- nificant difference was found (p=0.937). No statis- yngology of Dicle University School of Medicine. tically significant difference was found between the Patients were assessed by anamnesis and examina- pre- and post-operative means at the four frequencies tion and when necessary, by temporal computerized (p=0.712). tomography and diffusion magnetic resonance ima- Conclusion: In this study, we found that to reduce ging. Bone conduction thresholds at frequencies of complications relating to cholesteatoma, it might be 500, 1000, 2000, and 4000 Hz were determined by audiometric examination and they were compared necessary to completely remove the matrix especially before and after surgery. -
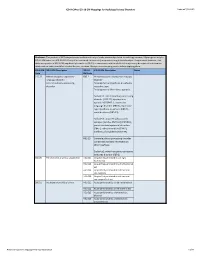
ICD-9/10 Mapping Spreadsheet
ICD-9-CM to ICD-10-CM Mappings for Audiology Related Disorders Updated 7/16/2015 Disclaimer: This product is NOT comprehensive and consists only of codes commonly related to audiology services. Mappings are only to ICD-10-CM codes, not ICD-10-PCS. Every effort was made to accurately map codes using detailed analysis. Keep in mind, however, that while many codes in ICD-9-CM map directly to codes in ICD-10, in some cases, additional clinical analysis may be required to determine which code or codes should be selected for your situation. Always review mapping results before applying them. ICD-9-CM ICD-9-CM Description ICD-10- ICD-10-CM Description Notes Code CM Code 315.32 Mixed receptive-expressive F80.2 Mixed receptive-expressive language language disorder disorder Central auditory processing Developmental dysphasia or aphasia, disorder receptive type Developmental Wernicke's aphasia Excludes1: central auditory processing disorder (H93.25), dysphasia or aphasia NOS (R47.-), expressive language disorder (F80.1), expressive type dysphasia or aphasia (F80.1), word deafness (H93.25) Excludes2: acquired aphasia with epilepsy [Landau-Kleffner] (G40.80-), pervasive developmental disorders (F84.-), selective mutism (F94.0), intellectual disabilities (F70-F79) H93.25 Central auditory processing disorder Congenital auditory imperception Word deafness Excludes1: mixed receptive-epxressive language disorder (F80.2) 380.00 Perichondritis of pinna, unspecified H61.001 Unspecified perichondritis of right external ear H61.002 Unspecified perichondritis -

Spectrum of Third Window Abnormalities: Semicircular Canal Dehiscence and Beyond
Published August 25, 2016 as 10.3174/ajnr.A4922 REVIEW ARTICLE Spectrum of Third Window Abnormalities: Semicircular Canal Dehiscence and Beyond X M.-L. Ho, X G. Moonis, X C.F. Halpin, and X H.D. Curtin ABSTRACT SUMMARY: Third window abnormalities are defects in the integrity of the bony structure of the inner ear, classically producing sound-/ pressure-induced vertigo (Tullio and Hennebert signs) and/or a low-frequency air-bone gap by audiometry. Specific anatomic defects include semicircular canal dehiscence, perilabyrinthine fistula, enlarged vestibular aqueduct, dehiscence of the scala vestibuli side of the cochlea, X-linked stapes gusher, and bone dyscrasias. We discuss these various entities and provide key examples from our institutional teaching file with a discussion of symptomatology, temporal bone CT, audiometry, and vestibular-evoked myogenic potentials. ABBREVIATIONS: EVAS ϭ enlarged vestibular aqueduct syndrome; SSCCD ϭ superior semicircular canal dehiscence hird window abnormalities are defects in the integrity of the ing effects decrease air conduction. The 2 physiologic windows Tbony structure of the inner ear, first described by Minor et al between the middle and inner ear are the oval window, which in 1998.1 In 2008, Merchant and Rosowski2 proposed a universal transmits vibrations from the auditory ossicles, and the round theory for the underlying mechanism of hearing loss accompany- window of the cochlea. With air conduction, there is physiologic ing these defects. Normal sound conduction is transmitted entrainment of the oval and round windows due to coupling by through the oval and round windows, which serve as fluid inter- incompressible perilymph. Pressure differences between the co- faces between air in the middle ear and perilymphatic fluid spaces chlear perilymphatic spaces activate hair cells and create the per- of the inner ear. -

The Coexistence of Labyrinthine Fistula and the Facial Canal Dehiscence
The Mediterranean Journal of Otology ORIGINAL ARTICLE Management of Labyrinthine Fistula and Accompanying Findings: The Coexistence of Labyrinthine Fistula and the Facial Canal Dehiscence Masoud Naderpour, Ghodrat Mohammadi, Najmeh Doostmohammadian Department of Otorhinolaryngology, Tabriz University of Medical science, Tabriz, Iran OBJECTIVE: To describe the audio-vestibular results of labyrinthine fistula surgery in patients with cholesteatoma. Correspondent Author: PATIENTS AND METHODS: Data of 185 patients who had undergone Chodrat Mohammadi Dept. Otorhnotaryngology Tabriz surgery for cholesteatoma between 2001 and 2007 were reviewed. University of Medical Sciences, Three-layer sealing was used for the management of fistula. Tabriz, ‹ran RESULTS: Twenty patients were found to have labyrinthine fistula, of which 11 (55%) were male and 9(45%) female. Fistula wase located in lateral Tel: + 98- 9141141619 semicircular canal in all cases. Correlation of labyrinthine fistula and facial E-mail: [email protected] nerve dehiscence was statistically significant. Follow up was done for 1- 6 year. Postoperatively, vertigo disappeared in 19 (95 %) patients. Submitted: 14 April 2008 Revised: 10 July 2008 Hearing remained unchanged in 18 (90 %) patients. Worsening in bone Accepted: 17 July 2008 conduction thresholds was observed in 2 (10 %) patients. Postoperative deafness did not occur. Mediterr J Otol 2008; 4: 132-137 CONCLUSION: Possibility of facial nerve dehiscence and tegmen defect should be considered in patients with labyrinthine fistula. Three-layer Copyright 2005 © The Mediterranean sealing may be a valuable technique in surgical treatment of labyrinthine Society of Otology and Audiology fistula, lowering the risk of cochleovestibular functions. 132 Management of labyrinthine fistula and accompanying findings Cholesteatoma is a pocket or cystic lesion consisting of Labyrinthine fistula (LF) is encountered during stratified squamous epithelium and proliferative keratin surgery for cholesteatoma with an average frequency within the temporal bone. -

Positive Perilymph Fistula Test with Semicircular Canal Dehiscence from Cholesteatoma
PRACTICE | CLINICAL IMAGES Positive perilymph fistula test with semicircular canal dehiscence from cholesteatoma Ming-Chih Hsieh MD, Chen Chi Wu MD PhD, Shih-Hao Wang MD n Cite as: CMAJ 2019 January 28;191:E104. doi: 10.1503/cmaj.180799 54-year-old man presented to our outpatient department with left-side hearing loss and tinnitus that had progressed for several years. The patient had vertigo with nausea, whichA was aggravated on applying pressure over the left external ear canal and tragus. Physical examination showed left-side tym- panic membrane retraction with a whitish mass at the epitympa- num, suggestive of cholesteatoma. Gently compressing the left-ear tragus induced apparently left-beating horizontal nystagmus (see video, Appendix 1, available at www.cmaj.ca/lookup/suppl/ doi:10.1503/cmaj.180799/-/DC1), consistent with a positive peri- lymph fistula test. Pure tone audiometry showed mixed-type hear- ing loss of 104 dB in the patient’s left ear and sensorineural hearing loss of 62 dB in his right. High-resolution computed tomography (CT) scan of the patient’s temporal bone showed a soft-tissue mass in his left middle ear and mastoid cavity with left lateral semicircular canal erosion (Appendix 2, available at www.cmaj.ca/lookup/suppl/ Figure 1: Microscopic view of left lateral semicircular canal dehiscence with doi:10.1503/cmaj.180799/-/DC1). These findings were compatible erosion of bony and membranous sections (arrows) in a 54-year-old man with with cholesteatoma with lateral semicircular canal dehiscence. cholesteatoma. Note: *the malleus handle; +second genu of the facial nerve; During surgery, we noted that the osseous and membranous por- dotted lines define the tympanic segment of the facial nerve. -

Radiology in Vertigo and Dizziness
10.5005/jp-journals-10003-1092 DeepakREVIEW Patkar ARTICLE et al Radiology in Vertigo and Dizziness Deepak Patkar, Girish Yevankar, Rashmi Parikh ABSTRACT • Cerebellopontine angle tumor Objective: Vertigo may or may not be associated with hearing • Cerebrovascular disease, such as transient ischemic loss. In addition, the imaging findings are often subtle. attack or stroke Underdiagnosis of cause of vertigo on imaging can lead to long • Migraine standing annoying symptom of vertigo. • Multiple sclerosis Conclusion: This article reviews the radiologic findings of • Other causes vertigo. After completing this article, the readers should have • Cervical vertigo an improved ability to diagnose the common causes of vertigo, • Drug-induced vertigo using the optimal imaging tools to achieve this goal. • Psychological Keywords: American College of Radiology, Magnetic The appropriate imaging modality depends upon the resonance imaging, Magnetic resonance angiogram. associated symptoms and is guided by ACR criterion as How to cite this article: Patkar D, Yevankar G, Parikh R. given in Table 1 (ACR Appropriateness Criteria)**.9 Radiology in Vertigo and Dizziness. Otorhinolaryngol Clin Int J 2012;4(2):86-92. Labyrinthitis Source of support: Nil Labyrinthitis is an inflammation of the membranous Conflict of interest: None declared labyrinth10 and can be divided according to etiology as follows: INTRODUCTION • Tympanogenic Imaging plays an important role in the diagnosis and • Post-traumatic treatment of the patients with vestibular abnormalities. • Meningogenic There are multiple imaging tools available for the • Hematogenic patients with vestibular and temporal bone abnormalities • Autoimmune viz computed tomography (CT) scan, magnetic resonance Prior to MRI, no imaging findings were described; imaging (MRI) including MR angiography and digital however, now enhancement of the labyrinth on postcontrast subtraction angiography. -

Surgical and Clinical Confirmation of Temporal Bone CT Findings In
ORIGINAL RESEARCH HEAD & NECK Surgical and Clinical Confirmation of Temporal Bone CT Findings in Patients with Otosclerosis with Failed Stapes Surgery J. Whetstone, A. Nguyen, A. Nguyen-Huynh, and B.E. Hamilton ABSTRACT BACKGROUND AND PURPOSE: Prior descriptions of imaging after failed stapes procedures for otosclerosis predated currently available CT technology and/or failed to assess commonly used metallic implants. The purpose of this study was to correlate temporal bone CT findings with clinically and intraoperatively determined causes of surgical failure. MATERIALS AND METHODS: All patients with otosclerosis undergoing stapedectomy between December 1999 and December 2010 were identified from a search of neurotology clinical records. Patients presenting because of failed stapes surgery and having temporal bone CT scans at the time of revision surgery or clinical evaluation were included. Imaging and clinical records were retrospectively evaluated by a medical student, radiology resident, and senior neuroradiologist. Stapes prosthesis complications and relevant anatomic CT findings were correlated to clinical and intraoperative findings. RESULTS: Twenty-two of 340 patients met inclusion criteria. Temporal bone CT findings were correlated to intraoperative findings in 17 of 22 patients and to clinical findings in 5 of 22 patients. Surgically confirmed abnormalities included 7 of 7 incus erosions, 3 of 6 piston re-sizings, 3 of 5 granulation tissues, 3 of 5 prosthesis disconnections, 3 of 4 obliterative otosclerosis, 2 of 2 oval window dislocations, and 1 labyrinthine ossificans. Clinically confirmed abnormalities included 2 cases each of superior semicircular canal dehiscence, and wrong piston size, and 1 each of piston disconnection, labyrinthine ossificans, and intravestibular footplate. CONCLUSIONS: CT evaluation in the setting of failed stapes surgery is challenging. -

WO 2017/178645 Al 19 October 2017 (19.10.2017) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2017/178645 Al 19 October 2017 (19.10.2017) P O P C T (51) International Patent Classification: BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, A61K 31/538 (2006.01) A61P 27/16 (2006.01) DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KH, KN, (21) International Application Number: KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, PCT/EP2017/059058 MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, (22) International Filing Date: NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, 14 April 2017 (14.04.2017) RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, (25) Filing Language: English ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 62/322,690 14 April 2016 (14.04.2016) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, 16180192.3 19 July 2016 (19.07.2016) EP TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, (71) Applicant: SENSORION [FR/FR]; 375 rue du Professeur DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, Joseph Blayac, 34080 Montpellier (FR). -

Management of the Labyrinthine Fistula in Chronic Otitis Media with Cholesteatoma by Z.Chafiki, M.Ait El Kerdoudi, S.Rouadi, R.L
Global Journal of Medical Research: J Dentistry & Otolaryngology Volume 16 Issue 1 Version 1.0 Year 2016 Type: Double Blind Peer Reviewed International Research Journal Publisher: Global Journals Inc. (USA) Online ISSN: 2249-4618 & Print ISSN: 0975-5888 Management of the Labyrinthine Fistula in Chronic Otitis Media with Cholesteatoma By Z.Chafiki, M.Ait el kerdoudi, S.Rouadi, R.L. Abada, M.Roubal & M.Mahtar Ibn Rochd University Hospital, Morocco Abstract- Labyrinthine fistula, defined by a destruction of the bony labyrinth is one of the most feared complications during the evolution of a chronic otitis media with cholesteatoma. Relatively common, it represents 4-12% cases of chronic otitis media with cholesteatoma and essentially occurs in the lateral semi-circular canal (70-80% of cases). The abnormal opening of the bony labyrinth in the middle ear runs the risk of recurrent labyrinthitis with dizziness, sensorineural hearing loss, purulent labyrinthitis and meningitis. Its treatment is surgical, however, an alteration of cochleovestibular function during surgery may occur. We propose in this development to review signs that suggest this complication and to establish a practical management of labyrinthine fistula in chronic otitis media with cholesteatoma. Keywords: cholesteatoma; fistula; labyrinthine; perilym-phatic; surgery. GJMR-J Classification: NLMC Code: WV 250 ManagementoftheLabyrinthineFistulainChronicOtitisMediawithCholesteatoma Strictly as per the compliance and regulations of: © 2016. Z.Chafiki, M.Ait el kerdoudi, S.Rouadi, R.L. Abada, M.Roubal & M.Mahtar. This is a research/review paper, distributed under the terms of the Creative Commons Attribution-Noncommercial 3.0 Unported License http://creativecommons.org/ licenses/by-nc/3.0/), permitting all non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited. -

Middle Ear Pathophysiology and Management Viewed from Pressure
PS 1 Middle Ear Pathophysiology and Management Viewed from Pressure-Regulation Function Haruo Takahashi Department of Otolaryngology – Head and Neck Surgery, Nagasaki University Graduate School of Biomedical Sciences It is well known that the middle ear (ME) pressure should be always kept atmospheric in order to maintain its efficient sound conductive function. For this purpose, there are two ME pressure-regulation systems; one is the eustachian tube (ET) and the other is transmucosal gas exchange, mainly in the mastoid. The ET function is known comparatively well, but the gas exchange function is not known so well. In my presentation, I first explain briefly the normal physiology of the gas exchange function, and then what happens to this important function under the condition of various ME diseases such as otitis media with effusion or cholesteatoma, and what happens to this function after ME surgery. Finally, I would like to propose the algorism of appropriate choice of ME surgical procedures according to pathophysiological conditions of the ME as well as to surgical intervention to the mastoid. PS 2 Otic Capsule Dehiscence Syndrome: Clinical Features, Comparison to Superior Semicircular Canal Dehiscence Syndrome and Longitudinal Cognitive Recovery with Surgery P. Ashley Wackym1, Carey D. Balaban2, Heather T. Mackay1, Dale M. Carter1, David A. Siker1, Scott J. Wood3 1Ear and Skull Base Center, Portland, OR 2University of Pittsburgh, Pittsburgh, PA 3Azusa Pacific University, Azusa, CA, USA Objective: (1) To longitudinally study two patient cohorts with superior semicircular canal dehiscence syndrome (SSCDS): one with radiographically confirmed superior semicircular canal dehiscence (SCD); and the other with no identified otic capsule dehiscence (no-iOCD). -

World's Largest Science, Technology & Medicine Open Access Book
We are IntechOpen, the world’s leading publisher of Open Access books Built by scientists, for scientists 5,400 133,000 165M Open access books available International authors and editors Downloads Our authors are among the 154 TOP 1% 12.2% Countries delivered to most cited scientists Contributors from top 500 universities Selection of our books indexed in the Book Citation Index in Web of Science™ Core Collection (BKCI) Interested in publishing with us? Contact [email protected] Numbers displayed above are based on latest data collected. For more information visit www.intechopen.com Chapter 3 Up to Date on Etiology and Epidemiology of Hearing Loss Larissa Vilela Pereira and Fayez Bahmad Jr. Additional information is available at the end of the chapter http://dx.doi.org/10.5772/61845 Abstract Deafness is one of the most common communication disorders in humans. Approximate‐ ly one out of every thousand infants is born with a significant hearing deficit. The preva‐ lence of hearing loss increases dramatically with age. By age 65 years, one out of three of us will suffer from hearing impairment sufficient to interfere with the understanding of speech. Hearing impairment is a very heterogeneous disorder with a wide range of caus‐ es. Worldwide, estimates from the World Health Organization are that hearing loss af‐ fects 538 million people. Hearing loss may be classified into three types: sensorineural, involving the inner ear, cochlea, or the auditory nerve; conductive, when the outer or middle ear structures fail to optimally capture, collect, or transmit sound to the cochlea; and mixed loss, which is a combination of conductive and sensorineural hearing loss. -

Thieme: Imaging of the Temporal Bone
fm 1/7/09 12:23 PM Page i Imaging of the Temporal Bone Fourth Edition fm 1/7/09 12:23 PM Page ii fm 1/7/09 12:23 PM Page iii Imaging of the Temporal Bone Fourth Edition Joel D. Swartz, MD President Germantown Imaging Associates Gladwyne, Pennsylvania Laurie A. Loevner, MD Professor of Radiology and Otorhinolaryngology—Head and Neck Surgery Department of Radiology Neuroradiology Section University of Pennsylvania School of Medicine and Health System Philadelphia, Pennsylvania Thieme New York • Stuttgart fm 1/7/09 12:23 PM Page iv Thieme Medical Publishers, Inc. 333 Seventh Ave. New York, NY 10001 Executive Editor: Timothy Hiscock Editorial Assistant: David Price Vice President, Production and Electronic Publishing: Anne T. Vinnicombe Production Editor: Heidi Pongratz, Maryland Composition Vice President, International Marketing and Sales: Cornelia Schulze Chief Financial Officer: Peter van Woerden President: Brian D. Scanlan Compositor: Thomson Digital Printer: The Maple-Vail Book Manufacturing Group Library of Congress Cataloging-in-Publication Data Imaging of the temporal bone / [edited by] Joel D. Swartz, Laurie A. Loevner.– 4th ed. p. ; cm. Rev. ed. of: Imaging of the temporal bone / Joel D. Swartz, H. Ric Harnsberger. 3rd ed. 1998. Includes bibliographical references and index. ISBN 978-1-58890-345-7 1. Temporal bone—Imaging. 2. Temporal bone—Diseases—Diagnosis. I. Swartz, Joel D. II. Loevner, Laurie A. [DNLM: 1. Temporal Bone—radiography. 2. Magnetic Resonance Imaging. 3. Temporal Bone—pathology. 4. Tomography, X-Ray Computed. WE 705 I31 2008] RF235.S93 2008 617'.514–dc22 2008026874 Copyright © 2009 by Thieme Medical Publishers, Inc.