University of Florida Thesis Or Dissertation
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Trimeresurus Sp
Trimeresurus sp. Copyright: Auszug aus Datenbank der Toxikologischen Abteilung der II. Medizinischen Klinik München; Toxinfo von Kleber JJ , Ganzert M, Zilker Th; Ausgabe 2002; erstellt Wagner Ph, Kleber JJ; Korthals Altes 1999 TOXIKOLOGIE: bei allen Arten von Trimeresurus Sp kommt es immer zu Lokalsymptomen bis Nekrose; zu rechnen ist mit außerdem mit Gerinnungsstörungen, Schocksymptomen T. ALBOLABRIS: massive Lokalsymptome, Gerinnungsstörung leicht bei 30%, stark bei 10% (16); Letalität in Thailand 3% (12) T. FLAVOVIRIDIS: vor Antiserum-Zeit 15% Letalität (12) starke Schwellung + Nekrosen, Schock, keine Gerinnungsstörungen bisher berichtet (11,12) T. GRAMINEUS: Schwellung; keine Nekrosen, keine Gerinnungsstörungen berichtet (1,14) T. KANBURIENSIS: Schwellung, Schock, Gerinnungsstörung (12) T. MUCROSQUAMATUS: Schwellung, Gerinnungsstörungen (12) T. POPEIORUM: Lokalsymptome; sehr geringe Gerinnungsstörung mit normalem Fibrinogen + Thrombo (15) T. PURPUREOMACULATUS: Schwellung, Nekrose, Gerinnungsstörung bis 40% (12,15) T. WAGLERI: Schwellung, Gerinnungsstörung (15) SYMPTOME: erste Vergiftungssymptome direkt nach dem Biß (sofortiger Schmerz, Schwellung entwickelt sich in den ersten 2-4 h) (2); meist starke Schwellung (häufig halbes bis ganzes Glied), bis ca. eine Woche anhaltend; Lymphangitis und schmerzhafte Lymphknotenschwellung (1,2,5,6) ; lokale subkutane Hämorrhagie, gelegentlich Blasenbildung und Hautnekrosen (1,2); bei T. flavoviridis auch Muskelnekrosen und Kompartmentsyndrom (3,12) MUND: lokal nach Giftaussaugen Schwellung an Lippe + Zunge bei T. albolabris (12) COR: selten Butdruckabfall, Schock (11,12); selten EKG-Veränderungen bei T. mucrosquamatus (12) LABOR: Thrombin ähnliche Aktivität führt zur Defibrinierung bis Verbrauchskoagulopathie mit Hypo- bis Afibrinogenämie, Thrombopenie (auch erst nach 12h Latenz) (1,2,4,13,16); Aktivierung der Fibrinolyse mit später Plasminerniedrigung (16); Leukozytose SONST: häufig Übelkeit, Erbrechen, Bauchschmerzen (11, 12); selten Nierenschädigung berichtet bei T. -

On Trimeresurus Sumatranus
See discussions, stats, and author profiles for this publication at: https://www.researchgate.net/publication/266262458 On Trimeresurus sumatranus (Raffles, 1822), with the designation of a neotype and the description of a new species of pitviper from Sumatra (Squamata: Viperidae: Crotalinae) Article in Amphibian and Reptile Conservation · September 2014 CITATIONS READS 4 360 3 authors, including: Gernot Vogel Irvan Sidik Independent Researcher Indonesian Institute of Sciences 102 PUBLICATIONS 1,139 CITATIONS 12 PUBLICATIONS 15 CITATIONS SEE PROFILE SEE PROFILE Some of the authors of this publication are also working on these related projects: Save Vietnam Biodiversity View project Systematics of the genus Pareas View project All content following this page was uploaded by Gernot Vogel on 01 October 2014. The user has requested enhancement of the downloaded file. Comparative dorsal view of the head of Trimeresurus gunaleni spec. nov. (left) and T. sumatranus (right). Left from above: male, female (holotype), male, all alive, from Sumatra Utara Province, Sumatra. Right: adult female alive from Bengkulu Province, Su- matra, adult male alive from Bengkulu Province, Sumatra, preserved female from Borneo. Photos: N. Maury. Amphib. Reptile Conserv. | amphibian-reptile-conservation.org (1) September 2014 | Volume 8 | Number 2 | e80 Copyright: © 2014 Vogel et al. This is an open-access article distributed under the terms of the Creative Commons Attribution–NonCommercial–NoDerivs 3.0 Unported License, Amphibian & Reptile Conservation which permits -

ICTV Virus Taxonomy Profile: Parvoviridae
ICTV VIRUS TAXONOMY PROFILES Cotmore et al., Journal of General Virology 2019;100:367–368 DOI 10.1099/jgv.0.001212 ICTV ICTV Virus Taxonomy Profile: Parvoviridae Susan F. Cotmore,1,* Mavis Agbandje-McKenna,2 Marta Canuti,3 John A. Chiorini,4 Anna-Maria Eis-Hubinger,5 Joseph Hughes,6 Mario Mietzsch,2 Sejal Modha,6 Mylene Ogliastro,7 Judit J. Penzes, 2 David J. Pintel,8 Jianming Qiu,9 Maria Soderlund-Venermo,10 Peter Tattersall,1,11 Peter Tijssen12 and ICTV Report Consortium Abstract Members of the family Parvoviridae are small, resilient, non-enveloped viruses with linear, single-stranded DNA genomes of 4–6 kb. Viruses in two subfamilies, the Parvovirinae and Densovirinae, are distinguished primarily by their respective ability to infect vertebrates (including humans) versus invertebrates. Being genetically limited, most parvoviruses require actively dividing host cells and are host and/or tissue specific. Some cause diseases, which range from subclinical to lethal. A few require co-infection with helper viruses from other families. This is a summary of the International Committee on Taxonomy of Viruses (ICTV) Report on the Parvoviridae, which is available at www.ictv.global/report/parvoviridae. Table 1. Characteristics of the family Parvoviridae Typical member: human parvovirus B19-J35 G1 (AY386330), species Primate erythroparvovirus 1, genus Erythroparvovirus, subfamily Parvovirinae Virion Small, non-enveloped, T=1 icosahedra, 23–28 nm in diameter Genome Linear, single-stranded DNA of 4–6 kb with short terminal hairpins Replication Rolling hairpin replication, a linear adaptation of rolling circle replication. Dynamic hairpin telomeres prime complementary strand and duplex strand-displacement synthesis; high mutation and recombination rates Translation Capped mRNAs; co-linear ORFs accessed by alternative splicing, non-consensus initiation or leaky scanning Host range Parvovirinae: mammals, birds, reptiles. -

Protoparvovirus Knocking at the Nuclear Door
viruses Review Protoparvovirus Knocking at the Nuclear Door Elina Mäntylä 1 ID , Michael Kann 2,3,4 and Maija Vihinen-Ranta 1,* 1 Department of Biological and Environmental Science and Nanoscience Center, University of Jyvaskyla, FI-40500 Jyvaskyla, Finland; elina.h.mantyla@jyu.fi 2 Laboratoire de Microbiologie Fondamentale et Pathogénicité, University of Bordeaux, UMR 5234, F-33076 Bordeaux, France; [email protected] 3 Centre national de la recherche scientifique (CNRS), Microbiologie Fondamentale et Pathogénicité, UMR 5234, F-33076 Bordeaux, France 4 Centre Hospitalier Universitaire de Bordeaux, Service de Virologie, F-33076 Bordeaux, France * Correspondence: maija.vihinen-ranta@jyu.fi; Tel.: +358-400-248-118 Received: 5 September 2017; Accepted: 29 September 2017; Published: 2 October 2017 Abstract: Protoparvoviruses target the nucleus due to their dependence on the cellular reproduction machinery during the replication and expression of their single-stranded DNA genome. In recent years, our understanding of the multistep process of the capsid nuclear import has improved, and led to the discovery of unique viral nuclear entry strategies. Preceded by endosomal transport, endosomal escape and microtubule-mediated movement to the vicinity of the nuclear envelope, the protoparvoviruses interact with the nuclear pore complexes. The capsids are transported actively across the nuclear pore complexes using nuclear import receptors. The nuclear import is sometimes accompanied by structural changes in the nuclear envelope, and is completed by intranuclear disassembly of capsids and chromatinization of the viral genome. This review discusses the nuclear import strategies of protoparvoviruses and describes its dynamics comprising active and passive movement, and directed and diffusive motion of capsids in the molecularly crowded environment of the cell. -

To the Minister of Infrastructure and Water Management Mrs S. Van Veldhoven-Van Der Meer P.O. Box 20901 2500 EX the Hague Dear M
To the Minister of Infrastructure and Water Management Mrs S. van Veldhoven-van der Meer P.O. Box 20901 2500 EX The Hague DATE 21 March 2018 REFERENCE CGM/180321-01 SUBJECT Advice on the pathogenicity classification of AAVs Dear Mrs Van Veldhoven, Further to a request for advice concerning the dossier IG 18-028_2.8-000 titled ‘Production of and activities involving recombinant adeno-associated virus (AAV) vectors with modified capsids and capsids of wild type AAV serotypes absent from Appendix 4’, submitted by Arthrogen B.V., COGEM hereby notifies you of the following: Summary: COGEM was asked to advise on the pathogenicity classification of five adeno-associated viruses (AAVs): AAV10, AAV11, AAV12, AAVrh10 and AAVpo1. More than 100 AAVs have been isolated from various hosts. AAVs can infect vertebrates, including humans (95% of people have at one time or another been exposed to an AAV). AAVs are dependent on a helper virus to complete their life cycle. In the absence of a helper virus, the AAV genome persists in a latent form in the cell nucleus. So far no mention has been made in the literature of disease symptoms caused by an infection with AAVs. Neither have there been any reports of pathogenicity of AAV10, 11, 12, AAVrh10 or AAVpo1. AAV vectors are regularly used in clinical studies. Given the above information, COGEM recommends assigning AAV10, AAV11, AAV12, AAVrh10 and AAVpo1 as non-pathogenic to pathogenicity class 1. Based on the ubiquity of AAVs and the lack of evidence of pathogenicity, COGEM recommends that all AAVs belonging to the species Adeno-associated dependoparvovirus A and Adeno-associated dependoparvovirus B should be assigned to pathogenicity class 1. -
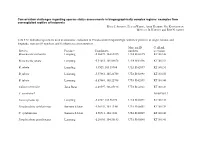
Conservation Challenges Regarding Species Status Assessments in Biogeographically Complex Regions: Examples from Overexploited Reptiles of Indonesia KYLE J
Conservation challenges regarding species status assessments in biogeographically complex regions: examples from overexploited reptiles of Indonesia KYLE J. SHANEY, ELIJAH WOSTL, AMIR HAMIDY, NIA KURNIAWAN MICHAEL B. HARVEY and ERIC N. SMITH TABLE S1 Individual specimens used in taxonomic evaluation of Pseudocalotes tympanistriga, with their province of origin, latitude and longitude, museum ID numbers, and GenBank accession numbers. Museum ID GenBank Species Province Coordinates numbers accession Bronchocela cristatella Lampung -5.36079, 104.63215 UTA R 62895 KT180148 Bronchocela jubata Lampung -5.54653, 105.04678 UTA R 62896 KT180152 B. jubata Lampung -5.5525, 105.18384 UTA R 62897 KT180151 B. jubata Lampung -5.57861, 105.22708 UTA R 62898 KT180150 B. jubata Lampung -5.57861, 105.22708 UTA R 62899 KT180146 Calotes versicolor Jawa Barat -6.49597, 106.85198 UTA R 62861 KT180149 C. versicolor* NC009683.1 Gonocephalus sp. Lampung -5.2787, 104.56198 UTA R 60571 KT180144 Pseudocalotes cybelidermus Sumatra Selatan -4.90149, 104.13401 UTA R 60551 KT180139 P. cybelidermus Sumatra Selatan -4.90711, 104.1348 UTA R 60549 KT180140 Pseudocalotes guttalineatus Lampung -5.28105, 104.56183 UTA R 60540 KT180141 P. guttalineatus Sumatra Selatan -4.90681, 104.13457 UTA R 60501 KT180142 Pseudocalotes rhammanotus Lampung -4.9394, 103.85292 MZB 10804 KT180147 Pseudocalotes species 4 Sumatra Barat -2.04294, 101.31129 MZB 13295 KT211019 Pseudocalotes tympanistriga Jawa Barat -6.74181, 107.0061 UTA R 60544 KT180143 P. tympanistriga Jawa Barat -6.74181, 107.0061 UTA R 60547 KT180145 Pogona vitticeps* AB166795.1 *Entry to GenBank by previous authors TABLE S2 Reptile species currently believed to occur Java and Sumatra, Indonesia, with IUCN Red List status, and certainty of occurrence. -

To the Minister for Infrastructure and Water Management Mrs C. Van Nieuwenhuizen-Wijbenga P.O. Box 20901 2500 EX the Hague Dear
To the Minister for Infrastructure and Water Management Mrs C. van Nieuwenhuizen-Wijbenga P.O. Box 20901 2500 EX The Hague DATUM 5 september 2019 KENMERK CGM/190905-01 ONDERWERP Advice 'Generic environmental risk assessment of clinical trials with AAV vectors' Dear Mrs Van Veldhoven, In the past COGEM has issued many advisory reports on gene therapy studies with AAV vectors. Armed with this knowledge, COGEM has drawn up a generic risk assessment for clinical applications of these vectors with the aim of streamlining the authorisation procedure for such trials. Summary: Hundreds of clinical trials have been carried out worldwide in which use was made of viral vectors derived from adeno-associated viruses (AAV). In recent years COGEM has published a large number of advisory reports on such trials, and in all these trials the risks to human health and the environment proved to be negligible. Drawing on these findings, COGEM has prepared a generic environmental risk assessment for clinical applications of AAV vectors. This generic environmental risk assessment can simplify and streamline the authorisation process. AAVs are non-pathogenic viruses that can only replicate in the presence of a helper virus. AAV vectors are stripped of all viral genes except the viral sequences at the ends of the genome, the inverted terminal repeats (ITRs). As a result, the vectors are unable to replicate even in the presence of a helper virus. COGEM therefore concludes that, given the characteristics of the AAV vectors used, the risks to human health and the environment posed by clinical trials with these vectors are negligible. -

Non-Invasive Surveys of Mammalian Viruses Using Environmental DNA
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.26.009993; this version posted March 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Title Non-invasive surveys of mammalian viruses using environmental DNA Highlights Environmental DNA (water and blood-sucking leeches) provided a non- invasive method of screening wildlife for viruses A comprehensive viral RNA oligonucleotide bait set was developed to capture known and unknown mammalian virus diversity Leech blood meal host determination and viruses identified were congruent Viruses determined from water correlated with known and observed species visiting the water sources In brief Alfano, Dayaram, et al. demonstrate that environmental DNA from southeast Asian leech bloodmeals and waterholes from Africa and Mongolia can be used as to detect viruses circulating in wildlife. These nucleic acid sources may represent an effective non-invasive resource for studying wildlife viral diversity and emerging viruses pre- emergence. bioRxiv preprint doi: https://doi.org/10.1101/2020.03.26.009993; this version posted March 29, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Non-invasive surveys of mammalian viruses using environmental DNA Niccolo Alfano1,2*, Anisha Dayaram3,4*, Jan Axtner1, Kyriakos Tsangaras5, Marie- Louise Kampmann1,6, Azlan Mohamed1,7, Seth T. -

Parvoviridae Hanafy .M.Madbouly Professor of Virology Faculty of Veterinary Medicine Beni-Suef University
Parvoviridae Hanafy .M.Madbouly Professor of Virology Faculty of Veterinary Medicine Beni-Suef University Definitions of the virus causative agents of several animal diseases. are most severe in fetuses and neonates. Parvovirus infections of the fetus or newborn during organogenesis may result in developmental defects. Replication of parvovirus is restricted in hemopoietic precursors, lymphocytes, and progenitor cells of intestinal mucosa of older animals. 2/38 Definitions of the virus cause acute infections for a few days, others persist for long periods in the feces of apparently robust host immune responses. Human Parvovirus B19 replication occurs only in human erythrocyte precursors The virus is found to survive in feces and other organic material such as soil for over 10 years. It survives extremely low and high temperatures. 3/38 Physical Properties of the virus . Family: Parvoviridae with two subfamilies . Parvovirinae. (vertebrate ). has 5 genera . Densovirinae. (invertebrate). Has3 genera .non-enveloped, 25 nm in diameter .Capsid: icosahederal. .Genome ssDNA Diseases caused by parvoviruses • As of 2014, there were no known human viruses in the remaining three recognized genera: • vi) Amdoparvovirus (e.g. Aleutian mink disease virus), • vii) Aveparvovirus (e.g. chicken parvovirus) and • viii) Copiparvovirus (e.g. bovine parvovirus 2). • Mouse parvovirus 1, however, causes no symptoms but can contaminate immunology experiments in biological research laboratories. • Porcine parvovirus causes a reproductive disease in swine known as SMEDI, which stands for stillbirth, mummification, embryonic death, and infertility. Diseases caused by parvoviruses • Many mammalian species sustain infection by multiple parvoviruses. • Feline panleukopenia is common in kittens and causes fever, low white blood cell count, diarrhea, and death. -

Evidence to Support Safe Return to Clinical Practice by Oral Health Professionals in Canada During the COVID-19 Pandemic: a Repo
Evidence to support safe return to clinical practice by oral health professionals in Canada during the COVID-19 pandemic: A report prepared for the Office of the Chief Dental Officer of Canada. November 2020 update This evidence synthesis was prepared for the Office of the Chief Dental Officer, based on a comprehensive review under contract by the following: Paul Allison, Faculty of Dentistry, McGill University Raphael Freitas de Souza, Faculty of Dentistry, McGill University Lilian Aboud, Faculty of Dentistry, McGill University Martin Morris, Library, McGill University November 30th, 2020 1 Contents Page Introduction 3 Project goal and specific objectives 3 Methods used to identify and include relevant literature 4 Report structure 5 Summary of update report 5 Report results a) Which patients are at greater risk of the consequences of COVID-19 and so 7 consideration should be given to delaying elective in-person oral health care? b) What are the signs and symptoms of COVID-19 that oral health professionals 9 should screen for prior to providing in-person health care? c) What evidence exists to support patient scheduling, waiting and other non- treatment management measures for in-person oral health care? 10 d) What evidence exists to support the use of various forms of personal protective equipment (PPE) while providing in-person oral health care? 13 e) What evidence exists to support the decontamination and re-use of PPE? 15 f) What evidence exists concerning the provision of aerosol-generating 16 procedures (AGP) as part of in-person -

An AAV-Based, Room-Temperature Stable, Single Dose COVID-19 Vaccine
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.05.422952; this version posted January 15, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. An AAV-based, room-temperature stable, single dose COVID-19 vaccine Nerea Zabaleta1,2,3,4, Wenlong Dai1,2,3,4, Urja Bhatt1,2,3,4, Jessica A Chichester5, Julio Sanmiguel1,2,3,4, Reynette Estelien1,2,3,4, Kristofer T Michalson5, Cheikh Diop1,2,3,4, Dawid Maciorowski1,2,3,4, Wenbin Qi6, Elissa Hudspeth7, Allison Cucalon1,2,3,4, Cecilia D Dyer5, M. Betina Pampena5,8, James J. Knox9, Regina C LaRocque10,11, Richelle C Charles10,11, Dan Li1,2,3,4, Maya Kim1,2,3,4, Abigail Sheridan1,2,3,4, Nadia Storm12, Rebecca I Johnson12, Jared Feldman13, Blake M Hauser13, Aisling Ryan1,2,3,4, Eric Zinn1,2,3,4, Dione T Kobayashi14, Ruchi Chauhan1,2,3,4, Marion McGlynn5, Edward T Ryan10,11,15, Aaron G Schmidt13,16, Brian Price17, Anna Honko12, Anthony Griffiths12, Sam Yaghmour6, Robert Hodge18, Michael R. Betts5,8, Mason W Freeman19, James M Wilson5, Luk H Vandenberghe1,2,3,4,* 1 Grousbeck Gene Therapy Center, Schepens Eye Research Institute, Mass Eye and Ear, Boston, Massachusetts, USA 2 Ocular Genomics Institute, Department of Ophthalmology, Harvard Medical School, Boston, Massachusetts, USA 3 The Broad Institute of Harvard and MIT, Cambridge, Massachusetts, USA 4 Harvard Stem Cell Institute, Harvard University, Cambridge, Massachusetts, USA 5Gene Therapy Program, Perelman School of Medicine, University -

A Phylogeny and Revised Classification of Squamata, Including 4161 Species of Lizards and Snakes
BMC Evolutionary Biology This Provisional PDF corresponds to the article as it appeared upon acceptance. Fully formatted PDF and full text (HTML) versions will be made available soon. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes BMC Evolutionary Biology 2013, 13:93 doi:10.1186/1471-2148-13-93 Robert Alexander Pyron ([email protected]) Frank T Burbrink ([email protected]) John J Wiens ([email protected]) ISSN 1471-2148 Article type Research article Submission date 30 January 2013 Acceptance date 19 March 2013 Publication date 29 April 2013 Article URL http://www.biomedcentral.com/1471-2148/13/93 Like all articles in BMC journals, this peer-reviewed article can be downloaded, printed and distributed freely for any purposes (see copyright notice below). Articles in BMC journals are listed in PubMed and archived at PubMed Central. For information about publishing your research in BMC journals or any BioMed Central journal, go to http://www.biomedcentral.com/info/authors/ © 2013 Pyron et al. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes Robert Alexander Pyron 1* * Corresponding author Email: [email protected] Frank T Burbrink 2,3 Email: [email protected] John J Wiens 4 Email: [email protected] 1 Department of Biological Sciences, The George Washington University, 2023 G St.