Clincal Trial Protocol
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(Rituxan®), Rituximab-Abbs (Truxima®), Rituximab-Pvvr (Ruxience®) Prior Authorization Drug Coverage Policy
1 Rituximab Products: Rituximab (Rituxan®), Rituximab-abbs (Truxima®), Rituximab-pvvr (Ruxience®) Prior Authorization Drug Coverage Policy Effective Date: 2/1/2021 Revision Date: n/a Review Date: 7/2/20 Lines of Business: Commercial Policy type: Prior Authorization This Drug Coverage Policy provides parameters for the coverage of rituximab (Rituxan®), rituximab-abbs (Truxima®), and rituximab-pvvr (Ruxience®). Consideration of medically necessary indications are based upon U.S. Food and Drug Administration (FDA) indications, recommended uses within the Centers of Medicare & Medicaid Services (CMS) five recognized compendia, including the National Comprehensive Cancer Network (NCCN) Drugs & Biologics Compendium (Category 1 or 2A recommendations), and peer-reviewed scientific literature eligible for coverage according to the CMS, Medicare Benefit Policy Manual, Chapter 15, section 50.4.5 titled, “Off- Label Use of Anti-Cancer Drugs and Biologics.” This policy evaluates whether the drug therapy is proven to be effective based on published evidence-based medicine. Drug Description1-3 Rituximab (Rituxan®), rituximab-abbs (Truxima®), and rituximab-pvvr (Ruxience®) are monoclonal antibodies that target the CD20 antigen expressed on the surface of pre-B and mature B- lymphocytes. Upon binding to cluster of differentiation (CD) 20, rituximab mediates B-cell lysis. Possible mechanisms of cell lysis include complement dependent cytotoxicity (CDC) and antibody dependent cell mediated cytotoxicity (ADCC). B cells are believed to play a role in the pathogenesis of rheumatoid arthritis (RA) and associated chronic synovitis. In this setting, B cells may be acting at multiple sites in the autoimmune/inflammatory process, including through production of rheumatoid factor (RF) and other autoantibodies, antigen presentation, T-cell activation, and/or proinflammatory cytokine production. -

Delivering Novel Targeted Therapies to Cancer Patients
NEXT-GENERATION RADIOIMMUNOTHERAPIES FOR NON-HODGKIN’S LYMPHOMA PATIENTS SEPTEMBER 2019 Nordic Nanovector ASA Kjelsåsveien 168 B, 0884 Oslo, Norway www.nordicnanovector.com IR contact: [email protected] Forward-looking statements This slide presentation contains certain forward-looking statements. These statements are based on management's current expectations and are subject to uncertainty and changes in circumstances, since they relate to events and depend on circumstances that will occur in the future and which, by their nature, will have an impact on Nordic Nanovector's business, financial condition and results of operations. The terms "anticipates", "assumes", "believes", "can", "could", "estimates", "expects", "forecasts", "intends", "may", "might", "plans", "should", "projects", "targets", "will", "would" or, in each case, their negative, or other variations or comparable terminology are used to identify forward looking statements. These forward-looking statements are not historic facts. There are a number of factors that could cause actual results and developments to differ materially from those expressed or implied in the forward-looking statements. Factors that could cause these differences include, but are not limited to, risks associated with implementation of Nordic Nanovector's strategy, risks and uncertainties associated with the development and/or approval of Nordic Nanovector's product candidates, ongoing and future clinical trials and expected trial results, the ability to commercialise Betalutin®, technology changes and new products in Nordic Nanovector's potential market and industry, Nordic Nanovector's freedom to operate (competitors patents) in respect of the products it develops, the ability to develop new products and enhance existing products, the impact of competition, changes in general economy and industry conditions, and legislative, regulatory and political factors. -

Unitedhealthcare Cancer Therapy Pathways Program Regimens
UnitedHealthcare Cancer Therapy Pathways Program Overview 2 Breast Cancer 3 Pancreatic Cancer 15 Melanoma 19 Colon/Rectal Cancer 23 Hepatobiliary Cancers 30 Lung Cancer, Small Cell 35 Lung Cancer, Non-Small Cell 40 Ovarian, Fallopian and Primary Peritoneal Cancer 56 Head and Neck Cancer 67 Multiple Myeloma 74 Lymphoma, Diffuse Large B-Cell 86 Lymphoma, Follicular 92 Lymphoma, Marginal Zone 97 Lymphoma, Mantle Cell 101 Change control 1 OVERVIEW Cancer Therapy Pathways Program With the rapid approval of new therapies, along with the rising cost of cancer care, pathways serve a critical role in the delivery of high-quality and high-value cancer treatments while reducing an unwarranted variation in care. The UnitedHealthcare Cancer Therapy Pathways Program aims to improve quality and value in cancer care by identifying anti-cancer regimens supported by evidence-based guidelines to help reduce total cost of care and improve outcomes. The program’s regimens are selected on the basis of clinical benefit (efficacy) and side-effect profile (toxicity). Among regimens with comparable efficacy and toxicity, additional consideration is given to the frequency of hospitalizations during therapy, duration of therapy, drug costs and total cost of care. Care decisions are between the physician and the patient The Cancer Therapy Pathways Program is not a substitute for the experience and judgment of a physician or other health care professional. Any clinician participating in the program must use independent medical judgment in the context of individual -

Pancreatic Cancer Treatment Using Na+/K+ Atpase
(19) & (11) EP 1 796 688 B1 (12) EUROPEAN PATENT SPECIFICATION (45) Date of publication and mention (51) Int Cl.: of the grant of the patent: A61K 31/704 (2006.01) A61P 35/00 (2006.01) 18.05.2011 Bulletin 2011/20 (86) International application number: (21) Application number: 05794315.1 PCT/US2005/031331 (22) Date of filing: 02.09.2005 (87) International publication number: WO 2006/028969 (16.03.2006 Gazette 2006/11) (54) PANCREATIC CANCER TREATMENT USING NA+/K+ ATPASE INHIBITORS PANKREASKREBSBEHANDLUNG MIT NA+/K+-ATPASE-HEMMERN TRAITEMENT DU CANCER DU PANCREAS AU MOYEN D’INHIBITEURS D’APTASE NA+/K+ (84) Designated Contracting States: • SHARMA, Ajay AT BE BG CH CY CZ DE DK EE ES FI FR GB GR Sudbury , MA 01776 (US) HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI SK TR (74) Representative: Tomkinson, Alexandra et al Bailey Walsh & Co (30) Priority: 02.09.2004 US 606684 P 5 York Place Leeds LS1 2SD (GB) (43) Date of publication of application: 20.06.2007 Bulletin 2007/25 (56) References cited: WO-A-00/45165 WO-A-03/099011 (73) Proprietor: Bionaut Pharmaceuticals Inc WO-A-2005/060951 WO-A-2005/077925 Stoneham, MA 02180 (US) US-A1- 2005 182 105 (72) Inventors: • KHODADOUST, Mehran - c/o BT Intern. Ltd. London EC4M 7SB (GB) Note: Within nine months of the publication of the mention of the grant of the European patent in the European Patent Bulletin, any person may give notice to the European Patent Office of opposition to that patent, in accordance with the Implementing Regulations. -
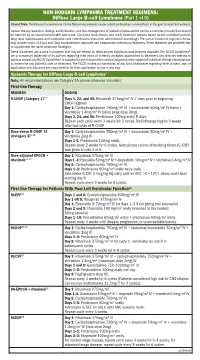
Diffuse Large B-Cell Lymphoma (Part 1 of 5)
NON-HODGKIN LYMPHOMA TREATMENT REGIMENS: Diffuse Large B-cell Lymphoma (Part 1 of 5) Clinical Trials: The National Comprehensive Cancer Network recommends cancer patient participation in clinical trials as the gold standard for treatment. Cancer therapy selection, dosing, administration, and the management of related adverse events can be a complex process that should be handled by an experienced health care team. Clinicians must choose and verify treatment options based on the individual patient; drug dose modifications and supportive care interventions should be administered accordingly. The cancer treatment regimens below may include both U.S. Food and Drug Administration-approved and unapproved indications/regimens. These regimens are provided only to supplement the latest treatment strategies. These Guidelines are a work in progress that may be refined as often as new significant data become available. The NCCN Guidelines® are a consensus statement of its authors regarding their views of currently accepted approaches to treatment. Any clinician seeking to apply or consult any NCCN Guidelines® is expected to use independent medical judgment in the context of individual clinical circumstances to determine any patient’s care or treatment. The NCCN makes no warranties of any kind whatsoever regarding their content, use, or application and disclaims any responsibility for their application or use in any way. Systemic Therapy for Diffuse Large B-cell Lymphoma1 Note: All recommendations are Category 2A unless otherwise indicated. First-line Therapy REGIMEN DOSING R-CHOP (Category 1)2–7 Days 1, 22, and 43: Rituximab 375mg/m2 IV 7 days prior to beginning CHOP regimen Day 1: Cyclophosphamide 750mg/m2 IV + doxorubicin 50mg/m2 IV bolus + vincristine 1.4mg/m2 IV bolus (max dose 2mg) Days 3, 24, and 45: Prednisone 100mg orally 5 days. -

De Novo Treatment of Diffuse Large B-Cell Lymphoma with Rituximab
Published Ahead of Print on November 12, 2013 as 10.1200/JCO.2013.49.7586 The latest version is at http://jco.ascopubs.org/cgi/doi/10.1200/JCO.2013.49.7586 JOURNAL OF CLINICAL ONCOLOGY ORIGINAL REPORT De Novo Treatment of Diffuse Large B-Cell Lymphoma With Rituximab, Cyclophosphamide, Vincristine, Gemcitabine, and Prednisolone in Patients With Cardiac Comorbidity: A United Kingdom National Cancer Research Institute Trial Paul A. Fields, William Townsend, Andrew Webb, Nicholas Counsell, Christopher Pocock, Paul Smith, Andrew Jack, Nadjet El-Mehidi, Peter W. Johnson, John Radford, David C. Linch, and David Cunnningham Paul A. Fields, Guys and St Thomas’ and King’s College Hospitals; William ABSTRACT Townsend, Nicholas Counsell, Paul Smith, and Nadjet El-Mehidi, Cancer Purpose Research UK and University College The treatment of patients with diffuse large B-cell lymphoma (DLBCL) with cardiac comorbidity is London Cancer Trials Centre; David C. problematic, because this group may not be able to receive anthracycline-containing chemoim- Linch, University College London Hospi- munotherapy. We designed a single-arm phase II multicenter trial of rituximab, gemcitabine, tal; David Cunnningham, Royal Marsden cyclophosphamide, vincristine, and prednisolone (R-GCVP) in patients considered unfit for Hospital, London; Andrew Webb, Royal anthracycline-containing chemoimmunotherapy because of cardiac comorbidity. Sussex Cancer Centre, Brighton; Chris- topher Pocock, Kent and Canterbury Patients and Methods Hospital, Kent; Andrew Jack, St James Sixty-one of 62 patients received R-GCVP, administered on day 1 with gemcitabine repeated on Hospital, Leeds; Peter W. Johnson, day 8 of a 21-day cycle. Median age was 76.5 years. All patients had advanced disease; 27 (43.5%) Southampton General Hospital, South- had left ventricular ejection fraction of Յ 50%, and 35 (56.5%) had borderline ejection fraction of ampton; and John Radford, Christie Ͼ Յ Hospital, Manchester, United Kingdom. -

Managing Relapsed and Refractory DLBCL
Managing relapsed and refractory DLBCL Andy Davies [email protected] Oxford Lymphoma Course June 2018 DLBCL is a curable disease Overall survival: All randomised 83.0% 82.0% HR=0.907 p=0.549 Real World Data Haematological Malignancies research Network 2017 Events occur early… Overall survival from Overall survival from diagnosis 2 years event free Maurer M J et al. JCO 2014;32:1066-1073 Outcomes of R-CHOP population 100% 90% 15-25% refractory 80% 5% PR patients 70% 20-30% relapses 60% 50% 40% 50-60% cured 30% 20% 10% 0% Coiffier and Sarkozy et al 2016 How can we identify those patients that may not respond well…? IPI Age greater than 60 years Stage III or IV disease Elevated serum LDH ECOG > 2 More than 1 extranodal site Age adjusted IPI Stage LDH Performance status Ziepert at al. J Clin Oncol 28:2373-2380. Primary Refractory Early relapse Late relapse Not eligible for HDT • Age Eligible for HDT • Co-morbidity consolidation • No response to reinduction • No stem cells at harvest Reinduction regimen Reinduction regimen (I hate ‘salvage’) Followed by HDT+PBSCT Primary Refractory Early relapse Late relapse Not eligible for HDT • Age Eligible for HDT • Co-morbidity consolidation • No response to reinduction • No stem cells at harvest Reinduction regimen Reinduction regimen (I hate ‘salvage’) Followed by HDT+PBSCT PARMA Trail (Phillip NEJM 1995) • PARMA study (n=215 aggressive relapsed disease) • 109 demonstrated chemosenstivity after DHAP x2: randomised DHAP x4 more or BEAC + ABMT • OS 53 vs 32% at 5 years (P=0.038) • Time to relapse (< or > 12 months most important prognostic factor, along with second line aaIPI and response to salvage PR vs CR Guglielmi et al, JCO 1998 PARMA TRIAL: ABMT vs DHAP (A) Actuarial PFS curves in responding early relapses (less than 12 months from initial diagnosis) according to treatment arm (ABMT versus DHAP). -

Bladder Cancer
Cancer Care Quality Program Treatment Pathways EFFECTIVE SEPTEMBER 27, 2021 LAST REVIEWED JULY 20, 2021 8600 West Bryn Mawr Avenue Appropriate.Safe.Affordable South Tower – Suite 800 Chicago, IL 60631 © 2021 AIM Specialty Health www.aimspecialtyhealth.com 9074-0921 Review and updates during 3rd quarter 2021 Bladder Cancer (Urothelial) • Modify existing clinical scenario, ‘Metastatic Disease | Second Line of Therapy (2nd Line)’ to ‘Metastatic Disease | Second and Subsequent Lines of Therapy (2nd Line +)’ o Added the following regimen, ‘Enfortumab Vedotin’ with footnote indicating that pathway status of this regimen requires prior therapy with platinum-based chemotherapy and PD-1/PD-L1 inhibitor Gastric, Esophageal, and Gastroesophageal Junction Cancer (Adenocarcinoma • Added the following regimen to the:, ‘Post-Operative Treatment’ clinical scenario: o Nivolumab (Opdivo) • Added the following regimen to the:, ‘Recurrent/Metastatic or Locally Advanced/Inoperable Disease | HER2 Negative | First Line of Therapy (1st Line)’ clinical scenario: o FOLFOX + nivolumab: fluorouracil (5FU), leucovorin, oxaliplatin, and nivolumab (Opdivo) (CPS > 5) • Modify existing clinical scenario, ‘Recurrent/Metastatic or Locally Advanced/Inoperable Disease | Second Line of Therapy (2nd Line)’ to ‘Recurrent/Metastatic or Locally Advanced/Inoperable Disease | Second and Subsequent Lines of Therapy (2nd Line +)’ o Added the following regimen, ‘Trastuzumab deruxtecan (Enhertu) – (HER2 Positive Only)’ Ovarian Cancer (Epithelial) • Removed the following regimen from the:, ‘Adjuvant Therapy | Stage IA/B (Grade 2 or 3) or IC (Grade 1-3)’ clinical scenario: o Carboplatin and dose dense paclitaxel Testicular (Germ Cell Tumors) Cancer • Modify existing clinical scenario, ‘Nonseminoma | Stage II-IIIA | Primary Therapy’ to ‘Nonseminoma | Stage IS-IIIA | Primary Therapy’ Note: Pathways are independent of specific health plan medical policy coverage criteria. -

Chemotherapy Drugs
CHEMOTHERAPY, DRUGS, AGENTS- This document contains three lists: 1) Individual drugs/agents 2) Known protocols 3) Old protocol descriptions Each table has the name of the drug, the code by which it is represented in the database, and the name by which this drug appears in the database if the drug can be known by more than one name. 1. Individual drugs/agents Drug Name (synonym) Code Name in database 2-CdA 63 2-CdA 4-demethoxydaunorubicin 18 Idarubicine 5- Fluorouracil 20 Fluorouracil 5FU 20 Fluorouracil 6-mercaptopurine 22 Mercaptapur6 6-MP 22 Mercaptapur6 Acridinyl anisidide 2 Amsacrine Actinomycin D 65 Dactinomycin Acyclovir 311 Acyclovir Adcetris 666 Brentuximab (Adcetris) Adriamycine 1 Adriamycine Adriblastin 1 Adriamycine Advagraf 52 Tacrolimus Agrylin 351 Anagrelide Aldesleukin 206 IL-2 Alemtuzumab 632 Campath (CD52, Alemtuzumab) Alexan 3 ARA-C / Cytarabine ALG 35 ALG Alkeran 21 Melphalan AMD-3100 36 Plerixafor Amethopterin 23 Methotrexate Amikin 326 Amikin AML like therapy 43 AML like therapy AMSA P-D 2 Amsacrine Amsacrine 2 Amsacrine Anagrelide 351 Anagrelide Analgesics 88 Analgesics Androgens 39 Androgens Anthracycline 57 Anthracycline Anti IL6 643 Anti IL6 anti rIL2 (CD25) 636 anti rIL2 (CD25) Anti tumour necrosis factor (TNF) 639 Anti tumour necrosis factor (TNF) Anti-GD2 644 Anti-GD2 antiLFA1 635 antiLFA1 Antilymphocyte globulin 35 ALG Anti-malarial 56 Anti-malarial Antimetabolites 93 Antimetabolites Antithymocyte globulin 34 ATG ARA-C 3 ARA-C Aranesp (Darbepoetin) 85 Aranesp (Darbepoetin) Aredia 82 Pamidronate Arranon 342 -

(12) United States Patent (10) Patent No.: US 9,603,952 B2 O'neill Et Al
US0096.03952B2 (12) United States Patent (10) Patent No.: US 9,603,952 B2 O'Neill et al. (45) Date of Patent: *Mar. 28, 2017 (54) TREATMENT OF METASTATIC TUMORS (56) References Cited (71) Applicant: MORPHOTEK, INC., Exton, PA (US) U.S. PATENT DOCUMENTS 4,444,744 A 4/1984 Goldenberg (72) Inventors: Alison O'Neill, Cambridge, MA (US); 5,051,364 A 9, 1991 Isacke et al. Douglas B. Jacoby, Wellesley, MA 5,212.290 A 5/1993 Vogelstein et al. (US); Abdellah Sentissi, Dover, MA 5,223,253 A 6/1993 Hall et al. (US); Kamala Kesavan, West Roxbury, 5,236,844 A 8, 1993 Basset et al. 5,314,992 A 5/1994 Guyre et al. MA (US); E. Michael Egan, Brookline, 5,591,829 A 1/1997 Matsushita MA (US) 5,626,862 A 5/1997 Brem et al. 5,688,773 A 11/1997 Chiocca et al. (73) Assignee: MORPHOTEK, INC., Exton, PA (US) 5,750,376 A 5/1998 Weiss et al. 5,756,340 A 5/1998 Hammock et al. (*) Notice: Subject to any disclaimer, the term of this 5,866,570 A 2/1999 Liang et al. patent is extended or adjusted under 35 5,905,027 A 5, 1999 Ullrich et al. 5,935,795 A 8, 1999 Lin et al. U.S.C. 154(b) by 0 days. 5,985,822 A 11/1999 Edelman et al. 6,028,174 A 2/2000 Ullrich et al. This patent is Subject to a terminal dis 6,130,101 A 10/2000 Mao et al. -

September 2020
September 2020 In This Issue Coverage Criteria Revised for Autonomic Nervous System Function Testing ........................................... 3 Coverage Guidelines Revised for Private Duty Nursing ............................................................................ 4 Coverage Guidelines Revised for Rituximab (Rituxan), Rituximab Biosimilars, and Rituximab and Hyaluronidase Human (Rituxan Hycela) ........................................................................................... 6 Contents .......................................................................................................................................................... 13 Policy C overage Guidelines Established for Pertuzumab, Trastuzumab, and Hyaluronidase-zzxf (Phesgo) Highmark Blue Shield has established new coverage criteria for pertuzumab, trastuzumab, and hyaluronidase-zzxf (Phesgo™) fixed dose subcutaneous formulation. The use of pertuzumab, trastuzumab, and hyaluronidase (Phesgo) may be considered medically when the presence of the HER2-overexpression is confirmed by the following: HER2-overexpression must be verified by ANY ONE of the following US Food and Drug Administration (FDA) approved diagnostic tests: • An immunohistochemical (IHC) assay with a result of 3+ (positive); or • A positive fluorescence in situ hybridization (FISH) test (ratio greater than 2.0); or • Single-probe in situ hybridization (ISH) test with average HER2 copy number 6.0 signals/cell or greater; or • Dual-probe ISH test HER2/CEP17 (chromosome enumeration probe -

(NCCN Guidelines®) B-Cell Lymphomas
NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) B-Cell Lymphomas Version 2.2018 — February 26, 2018 NCCN.org Continue Version 2.2018, 02/26/18 © National Comprehensive Cancer Network, Inc. 2018, All rights reserved. The NCCN Guidelines® and this illustration may not be reproduced in any form without the express written permission of NCCN®. Printed by Anton Kabakov on 3/5/2018 6:56:57 AM. For personal use only. Not approved for distribution. Copyright © 2018 National Comprehensive Cancer Network, Inc., All Rights Reserved. NCCN Guidelines Version 2.2018 Panel Members NCCN Guidelines Index Table of Contents B-Cell Lymphomas Discussion * Andrew D. Zelenetz, MD, PhD/Chair † Þ Beth Christian, MD † Ann S. LaCasce, MD † Memorial Sloan Kettering Cancer Center The Ohio State University Comprehensive Dana-Farber/Brigham and Women’s Cancer Center - James Cancer Hospital Cancer Center * Leo I. Gordon, MD/Co-Vice Chair ‡ ξ and Solove Research Institute Robert H. Lurie Comprehensive Cancer Amitkumar Mehta MD † ‡ Þ Center of Northwestern University Luis E. Fayad, MD ‡ Þ † University of Alabama at Birmingham The University of Texas Comprehensive Cancer Center Jeremy S. Abramson, MD † ‡ MD Anderson Cancer Center Massachusetts General Hospital Auayporn Nademanee, MD † ‡ ξ Cancer Center Martha J. Glenn, MD † ‡ Þ ξ City of Hope Comprehensive Cancer Center Huntsman Cancer Institute Ranjana H. Advani, MD † at the University of Utah Rachel Rabinovitch, MD § Stanford Cancer Institute University of Colorado Cancer Center Thomas M. Habermann, MD ‡ ξ C. Babis Andreadis, MD, MSCE ‡ † Mayo Clinic Cancer Center Nishitha Reddy, MD ‡ ξ UCSF Helen Diller Family Vanderbilt-Ingram Cancer Center Comprehensive Cancer Center Nancy Lee Harris, MD ≠ Erin Reid, MD ‡ Massachusetts General Hospital Cancer Center Nancy Bartlett, MD † UC San Diego Moores Cancer Center Siteman Cancer Center at Barnes- Francisco Hernandez-Ilizaliturri, MD † Stephen D.