Genetic Associations Between Voltage-Gated Calcium Channels (Vgccs) and Autism Spectrum Disorder (ASD)
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Mineralocorticoid Receptor Leads to Increased Expression of EGFR
www.nature.com/scientificreports OPEN The mineralocorticoid receptor leads to increased expression of EGFR and T‑type calcium channels that support HL‑1 cell hypertrophy Katharina Stroedecke1,2, Sandra Meinel1,2, Fritz Markwardt1, Udo Kloeckner1, Nicole Straetz1, Katja Quarch1, Barbara Schreier1, Michael Kopf1, Michael Gekle1 & Claudia Grossmann1* The EGF receptor (EGFR) has been extensively studied in tumor biology and recently a role in cardiovascular pathophysiology was suggested. The mineralocorticoid receptor (MR) is an important efector of the renin–angiotensin–aldosterone‑system and elicits pathophysiological efects in the cardiovascular system; however, the underlying molecular mechanisms are unclear. Our aim was to investigate the importance of EGFR for MR‑mediated cardiovascular pathophysiology because MR is known to induce EGFR expression. We identifed a SNP within the EGFR promoter that modulates MR‑induced EGFR expression. In RNA‑sequencing and qPCR experiments in heart tissue of EGFR KO and WT mice, changes in EGFR abundance led to diferential expression of cardiac ion channels, especially of the T‑type calcium channel CACNA1H. Accordingly, CACNA1H expression was increased in WT mice after in vivo MR activation by aldosterone but not in respective EGFR KO mice. Aldosterone‑ and EGF‑responsiveness of CACNA1H expression was confrmed in HL‑1 cells by Western blot and by measuring peak current density of T‑type calcium channels. Aldosterone‑induced CACNA1H protein expression could be abrogated by the EGFR inhibitor AG1478. Furthermore, inhibition of T‑type calcium channels with mibefradil or ML218 reduced diameter, volume and BNP levels in HL‑1 cells. In conclusion the MR regulates EGFR and CACNA1H expression, which has an efect on HL‑1 cell diameter, and the extent of this regulation seems to depend on the SNP‑216 (G/T) genotype. -

CACNB1 Antibody (C-Term) Blocking Peptide Synthetic Peptide Catalog # Bp16144b
10320 Camino Santa Fe, Suite G San Diego, CA 92121 Tel: 858.875.1900 Fax: 858.622.0609 CACNB1 Antibody (C-term) Blocking Peptide Synthetic peptide Catalog # BP16144b Specification CACNB1 Antibody (C-term) Blocking CACNB1 Antibody (C-term) Blocking Peptide - Peptide - Background Product Information The protein encoded by this gene belongs to Primary Accession Q02641 the calciumchannel beta subunit family. It plays an important role in thecalcium channel by modulating G protein inhibition, increasing CACNB1 Antibody (C-term) Blocking Peptide - Additional Information peakcalcium current, controlling the alpha-1 subunit membrane targetingand shifting the voltage dependence of activation and Gene ID 782 inactivation.Alternative splicing occurs at this locus and three transcriptvariants encoding Other Names three distinct isoforms have been identified. Voltage-dependent L-type calcium channel subunit beta-1, CAB1, Calcium channel CACNB1 Antibody (C-term) Blocking voltage-dependent subunit beta 1, CACNB1, Peptide - References CACNLB1 Format Jangsangthong, W., et al. Pflugers Arch. Peptides are lyophilized in a solid powder 459(3):399-411(2010)Olsen, J.V., et al. Cell format. Peptides can be reconstituted in 127(3):635-648(2006)Olsen, J.V., et al. Cell solution using the appropriate buffer as 127(3):635-648(2006)Lim, J., et al. Cell needed. 125(4):801-814(2006)Foell, J.D., et al. Physiol. Genomics 17(2):183-200(2004) Storage Maintain refrigerated at 2-8°C for up to 6 months. For long term storage store at -20°C. Precautions This product is for -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Noninvasive Sleep Monitoring in Large-Scale Screening of Knock-Out Mice
bioRxiv preprint doi: https://doi.org/10.1101/517680; this version posted January 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-ND 4.0 International license. Noninvasive sleep monitoring in large-scale screening of knock-out mice reveals novel sleep-related genes Shreyas S. Joshi1*, Mansi Sethi1*, Martin Striz1, Neil Cole2, James M. Denegre2, Jennifer Ryan2, Michael E. Lhamon3, Anuj Agarwal3, Steve Murray2, Robert E. Braun2, David W. Fardo4, Vivek Kumar2, Kevin D. Donohue3,5, Sridhar Sunderam6, Elissa J. Chesler2, Karen L. Svenson2, Bruce F. O'Hara1,3 1Dept. of Biology, University of Kentucky, Lexington, KY 40506, USA, 2The Jackson Laboratory, Bar Harbor, ME 04609, USA, 3Signal solutions, LLC, Lexington, KY 40503, USA, 4Dept. of Biostatistics, University of Kentucky, Lexington, KY 40536, USA, 5Dept. of Electrical and Computer Engineering, University of Kentucky, Lexington, KY 40506, USA. 6Dept. of Biomedical Engineering, University of Kentucky, Lexington, KY 40506, USA. *These authors contributed equally Address for correspondence and proofs: Shreyas S. Joshi, Ph.D. Dept. of Biology University of Kentucky 675 Rose Street 101 Morgan Building Lexington, KY 40506 U.S.A. Phone: (859) 257-2805 FAX: (859) 257-1717 Email: [email protected] Running title: Sleep changes in knockout mice bioRxiv preprint doi: https://doi.org/10.1101/517680; this version posted January 11, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Cdk15 Igfals Lingo4 Gjb3 Tpbg Lrrc38 Serpinf1 Apod Trp73 Lama4 Chrnd Col9a1col11a1col5a2 Fgl2 Pitx2 Col2a1 Col3a1 Lamb3 Col24a1
Bnc2 Wdr72 Ptchd1 Abtb2 Spag5 Zfp385a Trim17 Ier2 Il1rapl1 Tpd52l1 Fam20a Car8 Syt5 Plxnc1 Sema3e Ndrg4 Snph St6galnac5 Mcpt2 B3galt2 Sphkap Arhgap24 Prss34 Lhfpl2 Ermap Rnf165 Shroom1 Grm4 Mobp Dock2 Tmem9b Slc35d3 Otud7b Serpinb3a Sh3d19 Syt6 Zan Trim67 Clec18a Mcoln1 Tob1 Slc45a2 Pcdhb9 Pcdh17 Plscr1 Gpr143 Cela1 Frem1 Sema3f Lgi2 Igsf9 Fjx1 Cpne4 Adgb Depdc7 Gzmm C1qtnf5 Capn11 Sema3c H2-T22 Unc5c Sytl4 Galnt5 Sytl2 Arhgap11a Pcdha1 Cdh20 Slc35f2 Trim29 B3gnt5 Dock5 Trim9 Padi4 Pcdh19 Abi2 Cldn11 Slitrk1 Fam13a Nrgn Cpa4 Clmp Il1rap Trpm1 Fat4 Nexn Pmel Mmp15 Fat3 H2-M5 Prss38 Wdr41 Prtg Mlana Mettl22 Tnrc6b Cdh6 Sema3b Ptgfrn Cldn1 Cntn4 Bcl2a1b Capn6 Capn5 Pcdhb19 Tcf15 Bmf Rgs8 Tecrl Tyrp1 Rhot1 Rnf123 Cldn6 Adam9 Hlx Rilpl1 Disp1 Atcay Vwc2 Fat2 Srpx2 Cldn3 Unc13c Creb3l1 Rab39b Robo3 Gpnmb Bves Orai2 Slc22a2 Prss8 Cdh10 Scg3 Adam33 Nyx Dchs1 Chmp4c Syt9 Ap1m2 Megf10 Cthrc1 Penk Igsf9b Akap2 Ltbp3 Dnmbp Tff2 Pnoc Vldlr Cpa3 Snx18 Capn3 Btla Htr1b Gm17231 Pcdh9Rab27a Grm8 Cnih2 Scube2 Id2 Reep1 Cpeb3 Mmp16 Slc18b1 Snx33 Clcn5 Cckbr Pkp2 Drp2 Mapk8ip1 Lrrc3b Cxcl14 Zfhx3 Esrp1 Prx Dock3 Sec14l1 Prokr1 Pstpip2 Usp2 Cpvl Syn2 Ntn1 Ptger1 Rxfp3 Tyr Snap91 Htr1d Mtnr1a Gadd45g Mlph Drd4 Foxc2 Cldn4 Birc7 Cdh17 Twist2 Scnn1b Abcc4 Pkp1 Dlk2 Rab3b Amph Mreg Il33 Slit2 Hpse Micu1 Creb3l2 Dsp Lifr S1pr5 Krt15 Svep1 Ahnak Kcnh1 Sphk1 Vwce Clcf1 Ptch2 Pmp22 Sfrp1 Sema6a Lfng Hs3st5 Efcab1 Tlr5 Muc5acKalrn Vwa2 Fzd8 Lpar6 Bmp5 Slc16a9 Cacng4 Arvcf Igfbp2 Mrvi1 Dusp15 Krt5 Atp13a5 Dsg1a Kcnj14 Edn3Memo1 Ngef Prickle2 Cma1 Alx4 Bmp3 Blnk GastAgtr2 -

An Integrated Genomic Analysis of Gene-Function Correlation on Schizophrenia Susceptibility Genes
Journal of Human Genetics (2010) 55, 285–292 & 2010 The Japan Society of Human Genetics All rights reserved 1434-5161/10 $32.00 www.nature.com/jhg ORIGINAL ARTICLE An integrated genomic analysis of gene-function correlation on schizophrenia susceptibility genes Tearina T Chu and Ying Liu Schizophrenia is a highly complex inheritable disease characterized by numerous genetic susceptibility elements, each contributing a modest increase in risk for the disease. Although numerous linkage or association studies have identified a large set of schizophrenia-associated loci, many are controversial. In addition, only a small portion of these loci overlaps with the large cumulative pool of genes that have shown changes of expression in schizophrenia. Here, we applied a genomic gene-function approach to identify susceptibility loci that show direct effect on gene expression, leading to functional abnormalities in schizophrenia. We carried out an integrated analysis by cross-examination of the literature-based susceptibility loci with the schizophrenia-associated expression gene list obtained from our previous microarray study (Journal of Human Genetics (2009) 54: 665–75) using bioinformatic tools, followed by confirmation of gene expression changes using qPCR. We found nine genes (CHGB, SLC18A2, SLC25A27, ESD, C4A/C4B, TCP1, CHL1 and CTNNA2) demonstrate gene-function correlation involving: synapse and neurotransmission; energy metabolism and defense mechanisms; and molecular chaperone and cytoskeleton. Our findings further support the roles of these genes in genetic influence and functional consequences on the development of schizophrenia. It is interesting to note that four of the nine genes are located on chromosome 6, suggesting a special chromosomal vulnerability in schizophrenia. -

L-Type Cav 1.2 Calcium Channel-Α-1C Regulates Response to Rituximab in Diffuse Large B-Cell Lymphoma
Published OnlineFirst March 1, 2019; DOI: 10.1158/1078-0432.CCR-18-2146 Translational Cancer Mechanisms and Therapy Clinical Cancer Research L-Type Cav 1.2 Calcium Channel-a-1C Regulates Response to Rituximab in Diffuse Large B-Cell Lymphoma Jiu-Yang Zhang1, Pei-Pei Zhang1,Wen-Ping Zhou1, Jia-Yu Yu2, Zhi-Hua Yao1, Jun-Feng Chu1, Shu-Na Yao1, Cheng Wang2, Waseem Lone2, Qing-Xin Xia3, Jie Ma3, Shu-Jun Yang1, Kang-Dong Liu4,5, Zi-Gang Dong4, Yong-Jun Guo3, Lynette M. Smith6, Timothy W. McKeithan7, Wing C. Chan7, Javeed Iqbal2, and Yan-Yan Liu1 Abstract Purpose: One third of patients with diffuse large B-cell an independent prognostic factor for RCHOP resistance after lymphoma (DLBCL) succumb to the disease partly due to adjusting for International Prognostic Index, cell-of-origin rituximab resistance. Rituximab-induced calcium flux is an classification, and MYC/BCL2 double expression. Loss of important inducer of apoptotic cell death, and we investigated CACNA1C expression reduced rituximab-induced apoptosis the potential role of calcium channels in rituximab resistance. and tumor shrinkage. We further demonstrated direct inter- Experimental Design: The distinctive expression of calcium action of CACNA1C with CD20 and its role in CD20 stabi- channel members was compared between patients sensitive lization. Functional modulators of L-type calcium channel and resistant to rituximab, cyclophosphamide, vincristine, showed expected alteration in rituximab-induced apoptosis doxorubicin, prednisone (RCHOP) regimen. The observation and tumor suppression. Furthermore, we demonstrated that was further validated through mechanistic in vitro and in vivo CACNA1C expression was directly regulated by miR-363 studies using cell lines and patient-derived xenograft mouse whose high expression is associated with worse prognosis in models. -

An Advance About the Genetic Causes of Epilepsy
E3S Web of Conferences 271, 03068 (2021) https://doi.org/10.1051/e3sconf/202127103068 ICEPE 2021 An advance about the genetic causes of epilepsy Yu Sun1, a, *, †, Licheng Lu2, b, *, †, Lanxin Li3, c, *, †, Jingbo Wang4, d, *, † 1The School of Molecular and Cellular Biology, University of Illinois at Urbana-Champaign, Urbana, IL 61801-3633, US 2High School Affiliated to Shanghai Jiao Tong University, Shanghai, 200441, China 3Applied Biology program, University of British Columbia, Vancouver, V6r3b1, Canada 4School of Chemical Machinery and Safety, Dalian University of Technology, Dalian, 116023, China †These authors contributed equally. Abstract: Human hereditary epilepsy has been found related to ion channel mutations in voltage-gated channels (Na+, K+, Ca2+, Cl-), ligand gated channels (GABA receptors), and G-protein coupled receptors, such as Mass1. In addition, some transmembrane proteins or receptor genes, including PRRT2 and nAChR, and glucose transporter genes, such as GLUT1 and SLC2A1, are also about the onset of epilepsy. The discovery of these genetic defects has contributed greatly to our understanding of the pathology of epilepsy. This review focuses on introducing and summarizing epilepsy-associated genes and related findings in recent decades, pointing out related mutant genes that need to be further studied in the future. 1 Introduction Epilepsy is a neurological disorder characterized by 2 Malfunction of Ion channel epileptic seizures caused by abnormal brain activity. 1 in Functional variation in voltage or ligand-gated ion 100 (50 million people) people are affected by symptoms channel mutations is a major cause of idiopathic epilepsy, of this disorder worldwide, with men, young children, and especially in rare genetic forms. -

Ion Channels 3 1
r r r Cell Signalling Biology Michael J. Berridge Module 3 Ion Channels 3 1 Module 3 Ion Channels Synopsis Ion channels have two main signalling functions: either they can generate second messengers or they can function as effectors by responding to such messengers. Their role in signal generation is mainly centred on the Ca2 + signalling pathway, which has a large number of Ca2+ entry channels and internal Ca2+ release channels, both of which contribute to the generation of Ca2 + signals. Ion channels are also important effectors in that they mediate the action of different intracellular signalling pathways. There are a large number of K+ channels and many of these function in different + aspects of cell signalling. The voltage-dependent K (KV) channels regulate membrane potential and + excitability. The inward rectifier K (Kir) channel family has a number of important groups of channels + + such as the G protein-gated inward rectifier K (GIRK) channels and the ATP-sensitive K (KATP) + + channels. The two-pore domain K (K2P) channels are responsible for the large background K current. Some of the actions of Ca2 + are carried out by Ca2+-sensitive K+ channels and Ca2+-sensitive Cl − channels. The latter are members of a large group of chloride channels and transporters with multiple functions. There is a large family of ATP-binding cassette (ABC) transporters some of which have a signalling role in that they extrude signalling components from the cell. One of the ABC transporters is the cystic − − fibrosis transmembrane conductance regulator (CFTR) that conducts anions (Cl and HCO3 )and contributes to the osmotic gradient for the parallel flow of water in various transporting epithelia. -

Spatial Distribution of Leading Pacemaker Sites in the Normal, Intact Rat Sinoa
Supplementary Material Supplementary Figure 1: Spatial distribution of leading pacemaker sites in the normal, intact rat sinoatrial 5 nodes (SAN) plotted along a normalized y-axis between the superior vena cava (SVC) and inferior vena 6 cava (IVC) and a scaled x-axis in millimeters (n = 8). Colors correspond to treatment condition (black: 7 baseline, blue: 100 µM Acetylcholine (ACh), red: 500 nM Isoproterenol (ISO)). 1 Supplementary Figure 2: Spatial distribution of leading pacemaker sites before and after surgical 3 separation of the rat SAN (n = 5). Top: Intact SAN preparations with leading pacemaker sites plotted during 4 baseline conditions. Bottom: Surgically cut SAN preparations with leading pacemaker sites plotted during 5 baseline conditions (black) and exposure to pharmacological stimulation (blue: 100 µM ACh, red: 500 nM 6 ISO). 2 a &DUGLDFIoQChDQQHOV .FQM FOXVWHU &DFQDG &DFQDK *MD &DFQJ .FQLS .FQG .FQK .FQM &DFQDF &DFQE .FQM í $WSD .FQD .FQM í .FQN &DVT 5\U .FQM &DFQJ &DFQDG ,WSU 6FQD &DFQDG .FQQ &DFQDJ &DFQDG .FQD .FQT 6FQD 3OQ 6FQD +FQ *MD ,WSU 6FQE +FQ *MG .FQN .FQQ .FQN .FQD .FQE .FQQ +FQ &DFQDD &DFQE &DOP .FQM .FQD .FQN .FQG .FQN &DOP 6FQD .FQD 6FQE 6FQD 6FQD ,WSU +FQ 6FQD 5\U 6FQD 6FQE 6FQD .FQQ .FQH 6FQD &DFQE 6FQE .FQM FOXVWHU V6$1 L6$1 5$ /$ 3 b &DUGLDFReFHSWRUV $GUDF FOXVWHU $GUDD &DY &KUQE &KUP &KJD 0\O 3GHG &KUQD $GUE $GUDG &KUQE 5JV í 9LS $GUDE 7SP í 5JV 7QQF 3GHE 0\K $GUE *QDL $QN $GUDD $QN $QN &KUP $GUDE $NDS $WSE 5DPS &KUP 0\O &KUQD 6UF &KUQH $GUE &KUQD FOXVWHU V6$1 L6$1 5$ /$ 4 c 1HXURQDOPURWHLQV -

SUPPLEMENTARY APPENDIX Inflammation Regulates Long Non-Coding RNA-PTTG1-1:1 in Myeloid Leukemia
SUPPLEMENTARY APPENDIX Inflammation regulates long non-coding RNA-PTTG1-1:1 in myeloid leukemia Sébastien Chateauvieux, 1,2 Anthoula Gaigneaux, 1° Déborah Gérard, 1 Marion Orsini, 1 Franck Morceau, 1 Barbora Orlikova-Boyer, 1,2 Thomas Farge, 3,4 Christian Récher, 3,4,5 Jean-Emmanuel Sarry, 3,4 Mario Dicato 1 and Marc Diederich 2 °Current address: University of Luxembourg, Faculty of Science, Technology and Communication, Life Science Research Unit, Belvaux, Luxemburg. 1Laboratoire de Biologie Moléculaire et Cellulaire du Cancer, Hôpital Kirchberg, Luxembourg, Luxembourg; 2College of Pharmacy, Seoul National University, Gwanak-gu, Seoul, Korea; 3Cancer Research Center of Toulouse, UMR 1037 INSERM/ Université Toulouse III-Paul Sabatier, Toulouse, France; 4Université Toulouse III Paul Sabatier, Toulouse, France and 5Service d’Hématologie, Centre Hospitalier Universitaire de Toulouse, Institut Universitaire du Cancer de Toulouse Oncopôle, Toulouse, France Correspondence: MARC DIEDERICH - [email protected] doi:10.3324/haematol.2019.217281 Supplementary data Inflammation regulates long non-coding RNA-PTTG1-1:1 in myeloid leukemia Sébastien Chateauvieux1,2, Anthoula Gaigneaux1*, Déborah Gérard1, Marion Orsini1, Franck Morceau1, Barbora Orlikova-Boyer1,2, Thomas Farge3,4, Christian Récher3,4,5, Jean-Emmanuel Sarry3,4, Mario Dicato1 and Marc Diederich2 1 Laboratoire de Biologie Moléculaire et Cellulaire du Cancer, Hôpital Kirchberg, 9, rue Edward Steichen, 2540 Luxembourg, Luxemburg; 2 College of Pharmacy, Seoul National University, 1 Gwanak-ro, -
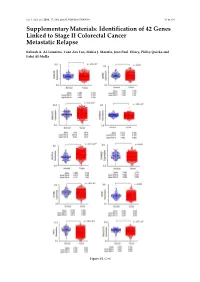
Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse
Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S1 of S16 Supplementary Materials: Identification of 42 Genes Linked to Stage II Colorectal Cancer Metastatic Relapse Rabeah A. Al-Temaimi, Tuan Zea Tan, Makia J. Marafie, Jean Paul Thiery, Philip Quirke and Fahd Al-Mulla Figure S1. Cont. Int. J. Mol. Sci. 2016, 17, 598; doi:10.3390/ijms17040598 S2 of S16 Figure S1. Mean expression levels of fourteen genes of significant association with CRC DFS and OS that are differentially expressed in normal colon compared to CRC tissues. Each dot represents a sample. Table S1. Copy number aberrations associated with poor disease-free survival and metastasis in early stage II CRC as predicted by STAC and SPPS combined methodologies with resident gene symbols. CN stands for copy number, whereas CNV is copy number variation. Region Cytoband % of CNV Count of Region Event Gene Symbols Length Location Overlap Genes chr1:113,025,076–113,199,133 174,057 p13.2 CN Loss 0.0 2 AKR7A2P1, SLC16A1 chr1:141,465,960–141,822,265 356,305 q12–q21.1 CN Gain 95.9 1 SRGAP2B MIR5087, LOC10013000 0, FLJ39739, LOC10028679 3, PPIAL4G, PPIAL4A, NBPF14, chr1:144,911,564–146,242,907 1,331,343 q21.1 CN Gain 99.6 16 NBPF15, NBPF16, PPIAL4E, NBPF16, PPIAL4D, PPIAL4F, LOC645166, LOC388692, FCGR1C chr1:177,209,428–177,226,812 17,384 q25.3 CN Gain 0.0 0 chr1:197,652,888–197,676,831 23,943 q32.1 CN Gain 0.0 1 KIF21B chr1:201,015,278–201,033,308 18,030 q32.1 CN Gain 0.0 1 PLEKHA6 chr1:201,289,154–201,298,247 9093 q32.1 CN Gain 0.0 0 chr1:216,820,186–217,043,421 223,235 q41 CN