The Effects of Atomoxetine on Cognitive Performace and Neuroplasticity After Traumatic Brain Injury
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

NINDS Custom Collection II
ACACETIN ACEBUTOLOL HYDROCHLORIDE ACECLIDINE HYDROCHLORIDE ACEMETACIN ACETAMINOPHEN ACETAMINOSALOL ACETANILIDE ACETARSOL ACETAZOLAMIDE ACETOHYDROXAMIC ACID ACETRIAZOIC ACID ACETYL TYROSINE ETHYL ESTER ACETYLCARNITINE ACETYLCHOLINE ACETYLCYSTEINE ACETYLGLUCOSAMINE ACETYLGLUTAMIC ACID ACETYL-L-LEUCINE ACETYLPHENYLALANINE ACETYLSEROTONIN ACETYLTRYPTOPHAN ACEXAMIC ACID ACIVICIN ACLACINOMYCIN A1 ACONITINE ACRIFLAVINIUM HYDROCHLORIDE ACRISORCIN ACTINONIN ACYCLOVIR ADENOSINE PHOSPHATE ADENOSINE ADRENALINE BITARTRATE AESCULIN AJMALINE AKLAVINE HYDROCHLORIDE ALANYL-dl-LEUCINE ALANYL-dl-PHENYLALANINE ALAPROCLATE ALBENDAZOLE ALBUTEROL ALEXIDINE HYDROCHLORIDE ALLANTOIN ALLOPURINOL ALMOTRIPTAN ALOIN ALPRENOLOL ALTRETAMINE ALVERINE CITRATE AMANTADINE HYDROCHLORIDE AMBROXOL HYDROCHLORIDE AMCINONIDE AMIKACIN SULFATE AMILORIDE HYDROCHLORIDE 3-AMINOBENZAMIDE gamma-AMINOBUTYRIC ACID AMINOCAPROIC ACID N- (2-AMINOETHYL)-4-CHLOROBENZAMIDE (RO-16-6491) AMINOGLUTETHIMIDE AMINOHIPPURIC ACID AMINOHYDROXYBUTYRIC ACID AMINOLEVULINIC ACID HYDROCHLORIDE AMINOPHENAZONE 3-AMINOPROPANESULPHONIC ACID AMINOPYRIDINE 9-AMINO-1,2,3,4-TETRAHYDROACRIDINE HYDROCHLORIDE AMINOTHIAZOLE AMIODARONE HYDROCHLORIDE AMIPRILOSE AMITRIPTYLINE HYDROCHLORIDE AMLODIPINE BESYLATE AMODIAQUINE DIHYDROCHLORIDE AMOXEPINE AMOXICILLIN AMPICILLIN SODIUM AMPROLIUM AMRINONE AMYGDALIN ANABASAMINE HYDROCHLORIDE ANABASINE HYDROCHLORIDE ANCITABINE HYDROCHLORIDE ANDROSTERONE SODIUM SULFATE ANIRACETAM ANISINDIONE ANISODAMINE ANISOMYCIN ANTAZOLINE PHOSPHATE ANTHRALIN ANTIMYCIN A (A1 shown) ANTIPYRINE APHYLLIC -

Aniracetam Reduces Glutamate Receptor Desensitization and Slows
Proc. Natl. Acad. Sci. USA Vol. 88, pp. 10936-10940, December 1991 Neurobiology Aniracetam reduces glutamate receptor desensitization and slows the decay of fast excitatory synaptic currents in the hippocampus (non-N-methyl-D-aspartate receptor/synapse) JEFFRY S. ISAACSON*t AND ROGER A. NICOLLtt *Physiology Graduate Program and the Departments of SPharmacology and tPhysiology, University of California, San Francisco, CA 94143-0450 Communicated by Floyd E. Bloom, September 16, 1991 ABSTRACT Aniracetam is a nootropic drug that has been and DL-2-amino-5-phosphonovaleric acid (50 ,uM) were shown to selectively enhance quisqualate receptor-mediated added to the medium to block y-aminobutyric acid type A responses inXenopus oocytes injected with brain mRNA and in (GABAA) receptors and NMDA receptors, respectively. In hippocampal pyramidal cells [Ito, I., Tanabe, S., Kohda, A. & the majority of experiments examining iontophoretic re- Sugiyama, H. (1990) J. Physiol. (London) 424, 533-544]. We sponses, tetrodotoxin (0.5-1 1uM) was included to block have used patch clamp recording techniques in hippocampal sodium-dependent action potentials. Currents were recorded slices to elucidate the mechanism for this selective action. We with an Axopatch 1B amplifier from neurons in the CA1 and find that aniracetam enhances glutamate-evoked currents in CA3 pyramidal cell layers and granule cell layer of the whole-cell recordings and, in outside-out patches, strongly dentate gyrus using the "blind" whole-cell recording tech- reduces glutamate receptor desensitization. In addition, nique (15, 16). Patch electrodes (tip diameter = 2 Ium) aniracetam selectively prolongs the time course and increases contained (in mM) either a CsF (110 CsF, 10 CsCl, 10 Hepes, the peak amplitude of fast synaptic currents. -

Neuroenhancement in Healthy Adults, Part I: Pharmaceutical
l Rese ca arc ni h li & C f B o i o l e Journal of a t h n Fond et al., J Clinic Res Bioeth 2015, 6:2 r i c u s o J DOI: 10.4172/2155-9627.1000213 ISSN: 2155-9627 Clinical Research & Bioethics Review Article Open Access Neuroenhancement in Healthy Adults, Part I: Pharmaceutical Cognitive Enhancement: A Systematic Review Fond G1,2*, Micoulaud-Franchi JA3, Macgregor A2, Richieri R3,4, Miot S5,6, Lopez R2, Abbar M7, Lancon C3 and Repantis D8 1Université Paris Est-Créteil, Psychiatry and Addiction Pole University Hospitals Henri Mondor, Inserm U955, Eq 15 Psychiatric Genetics, DHU Pe-psy, FondaMental Foundation, Scientific Cooperation Foundation Mental Health, National Network of Schizophrenia Expert Centers, F-94000, France 2Inserm 1061, University Psychiatry Service, University of Montpellier 1, CHU Montpellier F-34000, France 3POLE Academic Psychiatry, CHU Sainte-Marguerite, F-13274 Marseille, Cedex 09, France 4 Public Health Laboratory, Faculty of Medicine, EA 3279, F-13385 Marseille, Cedex 05, France 5Inserm U1061, Idiopathic Hypersomnia Narcolepsy National Reference Centre, Unit of sleep disorders, University of Montpellier 1, CHU Montpellier F-34000, Paris, France 6Inserm U952, CNRS UMR 7224, Pierre and Marie Curie University, F-75000, Paris, France 7CHU Carémeau, University of Nîmes, Nîmes, F-31000, France 8Department of Psychiatry, Charité-Universitätsmedizin Berlin, Campus Benjamin Franklin, Eschenallee 3, 14050 Berlin, Germany *Corresponding author: Dr. Guillaume Fond, Pole de Psychiatrie, Hôpital A. Chenevier, 40 rue de Mesly, Créteil F-94010, France, Tel: (33)178682372; Fax: (33)178682381; E-mail: [email protected] Received date: January 06, 2015, Accepted date: February 23, 2015, Published date: February 28, 2015 Copyright: © 2015 Fond G, et al. -

NIH Public Access Author Manuscript Addict Biol
NIH Public Access Author Manuscript Addict Biol. Author manuscript; available in PMC 2014 January 01. Published in final edited form as: Addict Biol. 2013 January ; 18(1): 54–65. doi:10.1111/adb.12000. Enhanced AMPA Receptor Activity Increases Operant Alcohol Self-administration and Cue-Induced Reinstatement $watermark-text $watermark-text $watermark-text Reginald Cannadya,b, Kristen R. Fishera, Brandon Duranta, Joyce Besheera,b,c, and Clyde W. Hodgea,b,c,d aBowles Center for Alcohol Studies, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599 bCurriculum in Neurobiology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599 cDepartment of Psychiatry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599 dDepartment of Pharmacology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599 Abstract Long-term alcohol exposure produces neuroadaptations that contribute to the progression of alcohol abuse disorders. Chronic alcohol consumption results in strengthened excitatory neurotransmission and increased AMPA receptor signaling in animal models. However, the mechanistic role of enhanced AMPA receptor activity in alcohol reinforcement and alcohol- seeking behavior remains unclear. This study examined the role of enhanced AMPA receptor function using the selective positive allosteric modulator, aniracetam, in modulating operant alcohol self-administration and cue-induced reinstatement. Male alcohol-preferring (P-) rats, trained to self-administer alcohol (15%, v/v) versus water were pretreated with aniracetam to assess effects on maintenance of alcohol self-administration. To determine reinforcer specificity, P-rats were trained to self-administer sucrose (0.8%, w/v) versus water, and effects of aniracetam were tested. -

Designing a Formulation of the Nootropic Drug Aniracetam Using 2-Hydroxypropyl-Β-Cyclodextrin Suitable for Parenteral Administration
pharmaceutics Article Designing a Formulation of the Nootropic Drug Aniracetam Using 2-Hydroxypropyl-β-Cyclodextrin Suitable for Parenteral Administration Sebastian D. Goldsmith and Arlene McDowell * School of Pharmacy, University of Otago, Dunedin 9054, New Zealand; [email protected] * Correspondence: [email protected]; Tel.: +64-3479-7145 Received: 17 October 2018; Accepted: 15 November 2018; Published: 18 November 2018 Abstract: The nootropic drug aniracetam is greatly limited in its application by low aqueous solubility and a poor oral bioavailability. The primary aim of this study was to design a parenteral formulation of aniracetam that can be administered intravenously. Complexation of aniracetam with 2-hydroxypropyl-β-cyclodextrin (HP-β-CD) was investigated as a strategy to enhance solubility. A phase solubility analysis was performed to quantify the extent of improvement. An 819% increase in the solubility of aniracetam was obtained, reaching 36.44 mg/mL. This marked increase enables aniracetam to exist in an aqueous solvent at levels sufficient for parenteral dosing. A stability test was then devised using a design of experiment approach. The aniracetam-HP-β-CD formulation was subjected to different relative humidity and temperature and cyclodextrin concentrations over a 12-week period. Key changes in FTIR vibrational frequencies suggest the benzene moiety of aniracetam was introduced into the hydrophobic cavity of HP-β-CD. These results are highly supportive of the formation of a predictable 1:1 molar stoichiometric inclusion complex, explaining the improvement seen in physiochemical properties of aniracetam following formulation with HP-β-CD. This novel formulation of aniracetam suitable for parenteral administration will have utility in future studies to further elucidate the pharmacokinetics of this drug. -

Mechanism of Positive Allosteric Modulators Acting on AMPA Receptors
The Journal of Neuroscience, September 28, 2005 • 25(39):9027–9036 • 9027 Cellular/Molecular Mechanism of Positive Allosteric Modulators Acting on AMPA Receptors Rongsheng Jin,1 Suzanne Clark,4 Autumn M. Weeks,4 Joshua T. Dudman,2 Eric Gouaux,1,3 and Kathryn M. Partin4 1Department of Biochemistry and Molecular Biophysics, 2Center for Neurobiology and Behavior, and 3Howard Hughes Medical Institute, Columbia University, New York, New York 10032, and 4Department of Biomedical Sciences, Division of Neuroscience, Colorado State University, Fort Collins, Colorado 80523 Ligand-gated ion channels involved in the modulation of synaptic strength are the AMPA, kainate, and NMDA glutamate receptors. Small molecules that potentiate AMPA receptor currents relieve cognitive deficits caused by neurodegenerative diseases such as Alzheimer’s disease and show promise in the treatment of depression. Previously, there has been limited understanding of the molecular mechanism of action for AMPA receptor potentiators. Here we present cocrystal structures of the glutamate receptor GluR2 S1S2 ligand-binding domain in complex with aniracetam [1-(4-methoxybenzoyl)-2-pyrrolidinone] or CX614 (pyrrolidino-1,3-oxazino benzo-1,4-dioxan-10- one), two AMPA receptor potentiators that preferentially slow AMPA receptor deactivation. Both potentiators bind within the dimer interface of the nondesensitized receptor at a common site located on the twofold axis of molecular symmetry. Importantly, the poten- tiator binding site is adjacent to the “hinge” in the ligand-binding core “clamshell” that undergoes conformational rearrangement after glutamatebinding.Usingrapidsolutionexchange,patch-clampelectrophysiologyexperiments,weshowthatpointmutationsofresidues that interact with potentiators in the cocrystal disrupt potentiator function. We suggest that the potentiators slow deactivation by stabilizing the clamshell in its closed-cleft, glutamate-bound conformation. -

Rat Animal Models for Screening Medications to Treat Alcohol Use Disorders
ACCEPTED MANUSCRIPT Selectively Bred Rats Page 1 of 75 Rat Animal Models for Screening Medications to Treat Alcohol Use Disorders Richard L. Bell*1, Sheketha R. Hauser1, Tiebing Liang2, Youssef Sari3, Antoinette Maldonado-Devincci4, and Zachary A. Rodd1 1Indiana University School of Medicine, Department of Psychiatry, Indianapolis, IN 46202, USA 2Indiana University School of Medicine, Department of Gastroenterology, Indianapolis, IN 46202, USA 3University of Toledo, Department of Pharmacology, Toledo, OH 43614, USA 4North Carolina A&T University, Department of Psychology, Greensboro, NC 27411, USA *Send correspondence to: Richard L. Bell, Ph.D.; Associate Professor; Department of Psychiatry; Indiana University School of Medicine; Neuroscience Research Building, NB300C; 320 West 15th Street; Indianapolis, IN 46202; e-mail: [email protected] MANUSCRIPT Key Words: alcohol use disorder; alcoholism; genetically predisposed; selectively bred; pharmacotherapy; family history positive; AA; HAD; P; msP; sP; UChB; WHP Chemical compounds studied in this article Ethanol (PubChem CID: 702); Acamprosate (PubChem CID: 71158); Baclofen (PubChem CID: 2284); Ceftriaxone (PubChem CID: 5479530); Fluoxetine (PubChem CID: 3386); Naltrexone (PubChem CID: 5360515); Prazosin (PubChem CID: 4893); Rolipram (PubChem CID: 5092); Topiramate (PubChem CID: 5284627); Varenicline (PubChem CID: 5310966) ACCEPTED _________________________________________________________________________________ This is the author's manuscript of the article published in final edited form as: Bell, R. L., Hauser, S. R., Liang, T., Sari, Y., Maldonado-Devincci, A., & Rodd, Z. A. (2017). Rat animal models for screening medications to treat alcohol use disorders. Neuropharmacology. https://doi.org/10.1016/j.neuropharm.2017.02.004 ACCEPTED MANUSCRIPT Selectively Bred Rats Page 2 of 75 The purpose of this review is to present animal research models that can be used to screen and/or repurpose medications for the treatment of alcohol abuse and dependence. -

Formulation Development Strategies
FORMULATION DEVELOPMENT STRATEGIES FOR ORAL EXTENDED RELEASE DOSAGE FORMS Dissertation zur Erlangung des akademischen Grades des Doktors der Naturwissenschaften (Dr. rer. nat.) eingereicht im Fachbereich Biologie, Chemie, Pharmazie der Freien Universität Berlin vorgelegt von ARAYA RAIWA aus Bangkok, Thailand May, 2011 1. Gutachter: Prof. Dr. Roland Bodmeier 2. Gutachter: Prof. Dr. Jürgen Siepmann Disputation am 9.Juni 2011 TO MY FAMILY ACKNOWLEDGEMENTS First and foremost, I wish to express my deepest gratitude to my supervisor, Prof. Dr. Roland Bodmeier for his professional guidance, helpful advices and encouragement. I am very grateful for his scientific and financial support and for providing me such an interesting topic. Furthermore, I am very thankful to him for the opportunity to support his editorial role in the European Journal of Pharmaceutical Sciences. I would like to thank Prof. Dr. Jürgen Siepmann for co-evaluating this thesis. Thanks are extended to Prof. Dr. Herbert Kolodziej, Prof. Dr. Johannes Peter Surmann and Dr. Martin Körber for serving as members of my thesis advisor committee. I am particular thankful to Dr. Andrei Dashevsky, Dr. Nantharat Pearnchob and Dr. Martin Körber for their very useful discussion; Dr. Burkhard Dickenhorst for evaluating parts of this thesis; Mrs. Angelika Schwarz for her assistance with administrative issues; Mr. Andreas Krause, Mrs. Eva Ewest and Mr. Stefan Walter for the prompt and diligent technical support. Sincere thanks are extended to Dr. Ildiko Terebesi, Dr. Burkhard Dickenhorst, Dr. Soravoot Rujivipat and Dr. Samar El-Samaligy for the friendly atmosphere in the lab My special thanks are owing to all members from the Kelchstrasse for their practical advice, enjoyable discussion and kindness throughout the years. -

Patent Application Publication ( 10 ) Pub . No . : US 2019 / 0192440 A1
US 20190192440A1 (19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2019 /0192440 A1 LI (43 ) Pub . Date : Jun . 27 , 2019 ( 54 ) ORAL DRUG DOSAGE FORM COMPRISING Publication Classification DRUG IN THE FORM OF NANOPARTICLES (51 ) Int . CI. A61K 9 / 20 (2006 .01 ) ( 71 ) Applicant: Triastek , Inc. , Nanjing ( CN ) A61K 9 /00 ( 2006 . 01) A61K 31/ 192 ( 2006 .01 ) (72 ) Inventor : Xiaoling LI , Dublin , CA (US ) A61K 9 / 24 ( 2006 .01 ) ( 52 ) U . S . CI. ( 21 ) Appl. No. : 16 /289 ,499 CPC . .. .. A61K 9 /2031 (2013 . 01 ) ; A61K 9 /0065 ( 22 ) Filed : Feb . 28 , 2019 (2013 .01 ) ; A61K 9 / 209 ( 2013 .01 ) ; A61K 9 /2027 ( 2013 .01 ) ; A61K 31/ 192 ( 2013. 01 ) ; Related U . S . Application Data A61K 9 /2072 ( 2013 .01 ) (63 ) Continuation of application No. 16 /028 ,305 , filed on Jul. 5 , 2018 , now Pat . No . 10 , 258 ,575 , which is a (57 ) ABSTRACT continuation of application No . 15 / 173 ,596 , filed on The present disclosure provides a stable solid pharmaceuti Jun . 3 , 2016 . cal dosage form for oral administration . The dosage form (60 ) Provisional application No . 62 /313 ,092 , filed on Mar. includes a substrate that forms at least one compartment and 24 , 2016 , provisional application No . 62 / 296 , 087 , a drug content loaded into the compartment. The dosage filed on Feb . 17 , 2016 , provisional application No . form is so designed that the active pharmaceutical ingredient 62 / 170, 645 , filed on Jun . 3 , 2015 . of the drug content is released in a controlled manner. Patent Application Publication Jun . 27 , 2019 Sheet 1 of 20 US 2019 /0192440 A1 FIG . -

Glutamate Receptors Within the Mesolimbic Dopamine System Mediate Alcohol Relapse Behavior
The Journal of Neuroscience, November 25, 2015 • 35(47):15523–15538 • 15523 Neurobiology of Disease Glutamate Receptors within the Mesolimbic Dopamine System Mediate Alcohol Relapse Behavior Manuela Eisenhardt,1,2 Sarah Leixner,1,2 Rafael Luja´n,3 Rainer Spanagel,1 and Ainhoa Bilbao1,2 1Institute of Psychopharmacology and 2Behavioral Genetics Research Group, Central Institute of Mental Health, Medical Faculty Mannheim/University of Heidelberg, J5, 68159 Mannheim, Germany, and 3Instituto de Investigacio´n en Discapacidades Neurolo´gicas, Departamento de Ciencias Me´dicas, Facultad de Medicina, Universidad Castilla-La Mancha, Campus Biosanitario, 02006 Albacete, Spain Glutamatergic input within the mesolimbic dopamine (DA) pathway plays a critical role in the development of addictive behavior. Although this is well established for some drugs of abuse, it is not known whether glutamate receptors within the mesolimbic system are involved in mediating the addictive properties of chronic alcohol use. Here we evaluated the contribution of mesolimbic NMDARs and AMPARs in mediating alcohol-seeking responses induced by environmental stimuli and relapse behavior using four inducible mutant mouse lines lacking the glutamate receptor genes Grin1 or Gria1 in either DA transporter (DAT) or D1R-expressing neurons. We first demonstrate the lack of GluN1 or GluA1 in either DAT- or D1R-expressing neurons in our mutant mouse lines by colocalization studies. We then show that GluN1 and GluA1 receptor subunits within these neuronal subpopulations mediate the alcohol deprivation effect, while having no impact on context- plus cue-induced reinstatement of alcohol-seeking behavior. We further validated these results pharmacologically by demonstrating similar reductions in the alcohol deprivation effect after infusion of the NMDAR antagonist me- mantine into the nucleus accumbens and ventral tegmental area of control mice, and a rescue of the mutant phenotype via pharmaco- logical potentiation of AMPAR activity using aniracetam. -

30Th Annual Meeting of Society for Neuroscience. New Drugs Affecting the Central Nervous System New Orleans, Louisiana, USA November 4–9, 2000
CNS Drug Reviews Vol. 7, No. 2, pp. 241–248 © 2001 Neva Press, Branford, Connecticut MEETING REPORT 30th Annual Meeting of Society for Neuroscience. New Drugs Affecting the Central Nervous System New Orleans, Louisiana, USA November 4–9, 2000 Alexander Scriabine Yale University School of Medicine, New Haven, CT, USA The 30th meeting of the Society for Neuroscience was held on November 4–9 in New Orleans, LA, USA. The meeting was attended by ~25,000 scientists and exhibitors. There were 874 slide or poster sessions, symposia or special lectures. Each session contained 10 to 20 presentations. This report covers only selected posters on new drugs affecting the central nervous system. NEUROPROTECTIVE DRUGS M. Cheng et al. (Centaur Pharmaceuticals, Inc., Sunnyvale, CA, USA) presented NXY-50, a novel free radical trapping agent, that was found to reduce infarct volume in rats with permanent focal ischemia induced by occlusion of middle cerebral artery (MCAO). The drug (30 or 60 mg/kg) was administered at 5 min after MCAO by i.v. bolus followed by infusion for 24 h (same dose per hour). The animals were sacrificed at 24 h after MCAO. The infarct volumes were estimated by histochemical staining. At 60 mg/kg the effect of the drug was statistically significant. J. Peeling et al. (Univ. of Mannitoba, Winnipeg, Canada, Memorial Univ., St. John’s, Newfoundland, Canada and AstraZeneca, Loughborough, UK and Södertalje, Sweden) studied NXY-059 in a rat model of hemorrhagic stroke. Intracerebral hemorrhage (ICH) was induced in rats by infusion of collagenase into SO Na 3 the right caudate nucleus. -
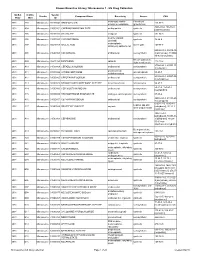
Known Bioactive Library: Microsource 1 - US Drug Collection
Known Bioactive Library: Microsource 1 - US Drug Collection ICCB-L ICCB-L Vendor Vendor Compound Name Bioactivity Source CAS Plate Well ID antifungal, inhibits Penicillium 2091 A03 Microsource 00200046 GRISEOFULVIN 126-07-8 mitosis in metaphase griseofulvum 3505-38-2, 486-16-8 2091 A04 Microsource 01500161 CARBINOXAMINE MALEATE antihistaminic synthetic [carbinoxamine] 2091 A05 Microsource 00200331 SALSALATE analgesic synthetic 552-94-3 muscle relaxant 2091 A06 Microsource 01500162 CARISOPRODOL synthetic 78-44-4 (skeletal) antineoplastic, 2091 A07 Microsource 00210369 GALLIC ACID insect galls 149-91-7 astringent, antibacterial 66592-87-8, 50370-12- 2091 A08 Microsource 01500163 CEFADROXIL antibacterial semisynthetic 2 [anhydrous], 119922- 89-9 [hemihydrate] Rheum palmatum, 2091 A09 Microsource 00211468 DANTHRON cathartic 117-10-2 Xyris semifuscata 27164-46-1, 25953-19- 2091 A10 Microsource 01500164 CEFAZOLIN SODIUM antibacterial semisynthetic 9 [cefazolin] glucocorticoid, 2091 A11 Microsource 00300024 HYDROCORTISONE adrenal glands 50-23-7 antiinflammatory 64485-93-4, 63527-52- 2091 A12 Microsource 01500165 CEFOTAXIME SODIUM antibacterial semisynthetic 6 [cefotaxime] 2091 A13 Microsource 00300029 DESOXYCORTICOSTERONE ACETATE mineralocorticoid adrenocortex 56-47-3 58-71-9, 153-61-7 2091 A14 Microsource 01500166 CEPHALOTHIN SODIUM antibacterial semisynthetic [cephalothin] 2091 A15 Microsource 00300034 TESTOSTERONE PROPIONATE androgen, antineoplastic semisynthetic 57-85-2 24356-60-3, 21593-23- 2091 A16 Microsource 01500167 CEPHAPIRIN SODIUM