Perfluorocarbons in the Global Atmosphere: Tetrafluoromethane
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Transport of Dangerous Goods
ST/SG/AC.10/1/Rev.16 (Vol.I) Recommendations on the TRANSPORT OF DANGEROUS GOODS Model Regulations Volume I Sixteenth revised edition UNITED NATIONS New York and Geneva, 2009 NOTE The designations employed and the presentation of the material in this publication do not imply the expression of any opinion whatsoever on the part of the Secretariat of the United Nations concerning the legal status of any country, territory, city or area, or of its authorities, or concerning the delimitation of its frontiers or boundaries. ST/SG/AC.10/1/Rev.16 (Vol.I) Copyright © United Nations, 2009 All rights reserved. No part of this publication may, for sales purposes, be reproduced, stored in a retrieval system or transmitted in any form or by any means, electronic, electrostatic, magnetic tape, mechanical, photocopying or otherwise, without prior permission in writing from the United Nations. UNITED NATIONS Sales No. E.09.VIII.2 ISBN 978-92-1-139136-7 (complete set of two volumes) ISSN 1014-5753 Volumes I and II not to be sold separately FOREWORD The Recommendations on the Transport of Dangerous Goods are addressed to governments and to the international organizations concerned with safety in the transport of dangerous goods. The first version, prepared by the United Nations Economic and Social Council's Committee of Experts on the Transport of Dangerous Goods, was published in 1956 (ST/ECA/43-E/CN.2/170). In response to developments in technology and the changing needs of users, they have been regularly amended and updated at succeeding sessions of the Committee of Experts pursuant to Resolution 645 G (XXIII) of 26 April 1957 of the Economic and Social Council and subsequent resolutions. -

Hexafluoroethane
Common Name: HEXAFLUOROETHANE CAS Number: 76-16-4 RTK Substance number: 0989 DOT Number: UN 2193 Date: May 1986 Revision: March 1999 ----------------------------------------------------------------------- ----------------------------------------------------------------------- HAZARD SUMMARY * Hexafluoroethane can affect you when breathed in. * If you think you are experiencing any work-related health * Contact can cause severe skin and eye irritation and burns problems, see a doctor trained to recognize occupational with possible eye damage. diseases. Take this Fact Sheet with you. * Breathing Hexafluoroethane can irritate the nose and throat. WORKPLACE EXPOSURE LIMITS * Breathing Hexafluoroethane can irritate the lungs causing No occupational exposure limits have been established for coughing and/or shortness of breath. Higher exposures Hexafluoroethane. This does not mean that this substance is can cause a build-up of fluid in the lungs (pulmonary not harmful. Safe work practices should always be followed. edema), a medical emergency, with severe shortness of breath. * Large amounts of Hexafluoroethane will decrease the * Exposure to the liquified gas can cause frostbite. amount of available Oxygen. Oxygen content should be * Hexafluoroethane can cause headache, nausea, fatigue, routinely tested to ensure that it is at least 19% by volume. mood changes, dizziness, unconsciousness and death. * Very high exposure can cause suffocation from lack of WAYS OF REDUCING EXPOSURE Oxygen. It may also cause the heart to beat irregularly * Where possible, enclose operations and use local exhaust (arrhythmia) or to stop, which can be fatal. ventilation at the site of chemical release. If local exhaust ventilation or enclosure is not used, respirators should be IDENTIFICATION worn. Hexafluoroethane is a colorless and odorless gas, or liquid * Wear protective work clothing. -

DECOMPOSITION PRODUCTS of FLUOROCARBON POLYMERS
criteria for a recommended standard . occupational exposure to DECOMPOSITION PRODUCTS of FLUOROCARBON POLYMERS U.S. DEPARTMENT OF HEALTH, EDUCATION, AND WELFARE criteria for a recommended standard... OCCUPATIONAL EXPOSURE TO DECOMPOSITION PRODUCTS of FLUOROCARBON POLYMERS U.S. DEPARTMENT OF HEALTH, EDUCATION, AND WELFARE Public Health Service Center for Disease Control National Institute for Occupational Safety and Health September 1977 For sale by the Superintendent of Documents, U.S. Government Printing Office, Washington, D.C. 20402 DHEW (NIOSH) Publication No. 77-193 PREFACE The Occupational Safety and Health Act of 1970 emphasizes the need for standards to protect the health and safety of workers exposed to an ever-increasing number of potential hazards at their workplace. The National Institute for Occupational Safety and Health has projected a formal system of research, with priorities determined on the basis of specified indices, to provide relevant data from which valid criteria for effective standards can be derived. Recommended standards for occupational exposure, which are the result of this work, are based on the health effects of exposure. The Secretary of Labor will weigh these recommendations along with other considerations such as feasibility and means of implementation in developing regulatory standards. It is intended to present successive reports as research and epidemiologic studies are completed and as sampling and analytical methods are developed. Criteria and standards will be reviewed periodically to ensure continuing protection of the worker. I am pleased to acknowledge the contributions to this report on the decomposition products of fluorocarbon polymers by members of the NIOSH staff and the valuable constructive comments by the Review Consultants on the Decomposition Products of Fluorocarbon Polymers and by Robert B. -

Special Applications
Special Applications Special Applications 04_Special Applications_04_SpecialApps 11/4/13 12:15 PM Page 1 Special Applications Contents Analytical Gases . .SA2 Certification Periods . .SA11 Certificates of Analysis . .SA11 Flame Ionization . .SA4 Air Toxics Monitoring . .SA12 Electron Capture . .SA4 Mobile Emissions Monitoring . .SA13 Leak Detection . .SA5 Airgas® LaserPlus™ Gases . .SA14 Nuclear Counter . .SA5 Refrigerant Gases . .SA16 Environmental Monitoring . .SA6 Process Chemical . .SA18 EPA Instrumental Test Methods . .SA7 Hydrocarbon Processing Gases . .SA20 Airgas® Safe-T-Cyl™ Lecture Bottle Replacement . .SA23 EPA Protocols and Traceability Standards . .SA8 Special Applications CEM Daily Calibration Standards . .SA9 Portable Cylinders . .SA24 Non-EPA Protocol Calibration Gases . .SA10 As the largest U.S. distributor of specialty gases, Airgas® has the expertise to prepare pure gases and mixtures designed for specific analytical, regulatory or industry processes. This section includes Analytical Gases, Environmental Monitoring Gases, Mobile Emissions Testing Gases. This section also includes refrigerants, process chemicals and other unique products that you’ll find with Airgas. Airgas Quality Policy The purpose of the Airgas Quality System is to continually improve our manufacturing and related processes to provide our customers with the highest product purity, consistency, and service. Among the Special Applications Gases are a full line of gases used in Environmental Monitoring, including EPA Protocols and Traceability Standards. -

Praxair Material Safety Data Sheet
Product: Hexafluoroethane P- 4670-G Date: December 2009 Praxair Material Safety Data Sheet 1. Chemical Product and Company Identification Product Name: Hexafluoroethane Trade Names: Halocarbon-116 (MSDS No. P-4670-G) Chemical Name: Hexafluoroethane Synonyms: : Halon-26, perfluoroethane, refrigerant gas R116 Chemical Family: Halogenated Alkane Product Grades: 3.6, 4.6 semiconductor process gas Telephone: Emergencies: 1-800-645-4633* Company Name: Praxair, Inc. CHEMTREC: 1-800-424-9300* 39 Old Ridgebury Road Routine: 1-800-PRAXAIR Danbury, CT 06810-5113 * Call emergency numbers 24 hours a day only for spills, leaks, fire, exposure, or accidents involving this product. For routine information, contact your supplier, Praxair sales representative, or call 1-800-PRAXAIR (1-800-772-9247). 2. Hazards Identification EMERGENCY OVERVIEW CAUTION! Liquid and gas under pressure. Can cause rapid suffocation. May cause frostbite. May cause dizziness and drowsiness. Self-contained breathing apparatus may be required by rescue workers. Under ambient conditions, this is a colorless, odorless gas at normal temperature and pressure. OSHA REGULATORY STATUS: This material is considered hazardous by the OSHA Hazard Communications Standard (29 CFR 1910.1200). POTENTIAL HEALTH EFFECTS: Effects of a Single (Acute) Overexposure Inhalation. Asphyxiant. High concentrations may cause dizziness, nausea, vomiting, disorientation, confusion, incoordination, and narcosis. Very high concentrations may cause suffocation. Lack of oxygen can kill. Skin Contact. Liquid hexafluoroethane may cause frostbite; harmful amounts may be absorbed if skin contact is prolonged or widespread. Swallowing. An unlikely route of exposure, but frostbite of the lips and mouth may result from contact with the liquid. Eye Contact. Liquid may cause severe corneal injury. -

Tetrafluoroethylene
TETRAFLUOROETHYLENE Tetrafluoroethylene was reviewed previously by the Working Group in 1979, 1987, and 1998 (IARC, 1979, 1987, 1999). New data have since become available, and these have been incor- porated, and taken into consideration in the present evaluation. 1. Exposure Data 1.1.3 Chemical and physical properties of the pure substance 1.1 Identification of the agent From IFA (2014), unless otherwise indicated 1.1.1 Nomenclature Description: Colourless gas, odourless or sometimes described as having a faint Chem. Abstr. Serv. Reg. No.: 116-14-3 sweetish odour; extremely flammable Chem. Abstr. Serv. Name: Tetrafluoroethylene Boiling point: −75.63 °C IUPAC Systematic Name: Melting point: −131.15 °C (HSDB, 2014) 1,1,2,2-Tetrafluoroethene Density: 4216 kg/m3 at 15 °C at 1 bar Synonyms: Perfluoroethylene, Perfluoroethene, Solubility: Slightly soluble in water, 159 mg/L Ethylene tetrafluoro-, tetrafluoroethene at 25 °C (HSDB, 2014) Vapour pressure: 2947 kPa and 20 °C 1.1.2 Structural and molecular formulae, and Stability: Decomposes into fluorine and fluo- relative molecular mass rine compounds when heated (HSDB, 2014) Reactivity: A terpene inhibitor (limonene) is F F generally added to the monomer to prevent CC spontaneous polymerization. F F Risk of explosion in contact with air or in the absence of air at elevated temperatures and/or Molecular formula: C2F4 pressures (> 600 °C and 100 kPa). The stabilized Relative molecular mass: 100.01 monomer is flammable in air if ignited (flamma- bility limits: lower, 11%; upper, 60%) producing soot and carbon tetrafluoride Babenko( et al., 1993; HSDB, 2014). Incompatible with polymerization catalysts and peroxides. -

Section 2. Hazards Identification OSHA/HCS Status : This Material Is Considered Hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200)
SAFETY DATA SHEET Halocarbon R-508A Section 1. Identification GHS product identifier : Halocarbon R-508A Other means of : Blend of Hexafluoroethane/Trifluoromethane; R116/R23, PFC116/HFC23 identification Product type : Gas. Product use : Synthetic/Analytical chemistry. Synonym : Blend of Hexafluoroethane/Trifluoromethane; R116/R23, PFC116/HFC23 SDS # : 008070 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : GASES UNDER PRESSURE - Compressed gas substance or mixture GHS label elements Hazard pictograms : Signal word : Warning Hazard statements : Contains gas under pressure; may explode if heated. May displace oxygen and cause rapid suffocation. Precautionary statements General : Read and follow all Safety Data Sheets (SDS’S) before use. Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Close valve after each use and when empty. Use equipment rated for cylinder pressure. Do not open valve until connected to equipment prepared for use. Use a back flow preventative device in the piping. Use only equipment of compatible materials of construction. Prevention : Not applicable. Response : Not applicable. Storage : Protect from sunlight. Store in a well-ventilated place. Disposal : Not applicable. Hazards not otherwise : In addition to any other important health or physical hazards, this product may displace classified oxygen and cause rapid suffocation. Date of issue/Date of revision : 8/17/2018 Date of previous issue : No previous validation Version : 1 1/11 Halocarbon R-508A Section 3. -
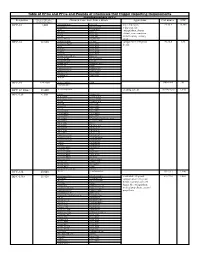
Table of Hfcs and Pfcs and Pounds of Chemicals That Trigger
Table of HFCs and PFCs and Pounds of Chemicals that Trigger Reporting Requirements Hydrofluorocarbons (HFCs) Designation Pounds of Substance = Chemical Name, Trade Names, Blends Applications CAS number GWP 10,000 metric tons Ce HFC-23 1,480 trifluoromethane Suva 95 low temperature 75-46-7 14,800 fluoroform Genetron 503 refrigerant, fire R-23 Genetron 23 extinguishant, plasma Freon 23 Klea 23 FE-13 Klea 508 etchant, semi-conductor FE-36 Klea 5R3 manufacturing cleaning Forane FX 220 NARM 503 agent HFC-32 32,660 difluoromethane R-407C component in refrigerant 75-10-5 675 methylene fluoride Klea 407A blends R-32 Klea 407B R-410A Klea 407C R-407C Klea 407D Freon 32 Klea 410A Forane 410A (AZ-20) Klea 32 Forane FX 40 Genetron 407C Forane FX 220 AZ-20 Forane 407C EcoloAce 407c Forane 32 HX4 Asahiklin SA-39 Solkane 407C Asahiklin SA-45 Solkane 410 Meforex 98 Suva 9000 Meforex 32 Suva 9100 R-410A HFC-41 227,280 methyl fluoride R-41 593-53-3 97 fluoromethane HFC-43-10mee 13,440 decafluoropentane cleaning solvent 138495-42-8 1,640 HFC-125 6,300 ethane Forane 507 pentafluoro Forane FX10 pentafluoroethane Forane FX40 R-125 Forane FX70 FC-125 Suva 125 Freon 125 Suva 9000 FE-25 Suva 9100 Arcton 402A Suva HP62 Arcton 402B Suva HP80 Arcton 408A Suva HP81 Klea 404A Cooltop R-134a Replacement Klea 407A EcoloAce 404a Klea 407B Di 44 Klea 407C Meforex 125 Klea 407D Meforex 55 Klea 410A Meforex 57 Klea 507A Meforex 98 Asahiklin SA-28 ISCEON 29 Asahiklin SA-39 ISCEON 404A Asahiklin SA-45 ISCEON 507 AZ-20 ISCEON 59 AZ-50 ISCEON 79 Genetron 404A ISCEON 89 Genetron -

3017.0001 the Fluorides of the Actinide Elements
ADVANCES IN FLUORINE CHEMISTRY VOLUME 2 EDITOR8 M. STACEY, F.R.S. A,Iaso~t Professor and tIead qf Deparlment of Chemistry, University of Birmingham J. C. TATLOW, Ph.D., D.Sc. Professor of Organic Chemistry, University of Birmingham A. G. SHARPE, M.A., Ph.D. Universi!~ Lecturer in Chemisto,, Cambridge WASHINGTON BUTTERWORTHS 1961 3017.0001 THE FLUORIDES OF THE ACTINIDE ELEMENTS Templeton, D. H. and Dauben, C. H. J. Amer. Chem. Soc. 1953, 75, 4560 Carniglia, S. C. and Cunningham, B. B. J. Amer. Chem. Soc. 1955, 77, 1451 Westrum, E. F. and Eyring, L. J. Amer. Chem. Soc. 1951, 73, 3396 Feay, D. C. Some Chemical Properties of Curium, Thesis, University of California. See also U.S.A.E.C. Report, UCRL-2547, 1954 Eyring, L., Cunningham, B. B. and Lohr, H. R. J. Amer. CAem. Soc. 1952, 74, 1186 Yakovlev, G. N. and Kosyakov, V. N. Proc. 2nd U.N. Co~fc. Pc’a@d Uses Atomic Energy, Geneva, 1958, paper 2127, 28, 373, 1958 Asprey, L. B. and Keenan, T. K. J. Inorg. Nuclear Chem. 1958, 7, 27 Asprey, L. B., Ellinger, F. H. and Zacl~ariasen, W. H. J. Amer. Chem. Soc. 1954, 76, 5235 Wallmann, J. C., Crane, W. W. T. and Cunningttam, B. 13. J. Amer. Chem. Soc. 1951, "/3, 493 Asprey, L. B., Ellinger, F. H., Fried, S. and Zachariasen, W. H. J. Ame~’. CheDz. Soc. 1957, ~, 5825 Asprey, L. B. and Ellinger, F. H. U.S.A.E.C. Report, AECD-3627, declassified 1954 Crane, W. W. T. U.S.A.E.C. -

3.3 Greenhouse Gas Emissions Final EIR | Spring Street Business Park Project
3.3 Greenhouse Gas Emissions Final EIR | Spring Street Business Park Project 3.3 Greenhouse Gas Emissions 3.3.1 Overview This section describes the existing air quality conditions and applicable laws and regulations associated with air quality, as well as an analysis of the potential effects resulting from implementation of the proposed project. Information contained in this section is summarized from the Air Quality and Greenhouse Gas Technical Memorandum (Appendix B). 3.3.2 Environmental Setting Climate change refers to long-term changes in temperature, precipitation, wind patterns, and other elements of the earth's climate system. An ever-increasing body of scientific research attributes these climatological changes to GHG emissions, particularly those generated from the production and use of fossil fuels. While climate change has been a concern for several decades, the establishment of the Intergovernmental Panel on Climate Change by the United Nations and World Meteorological Organization in 1988 has led to increased efforts devoted to GHG emissions reduction and climate change research and policy. These efforts are primarily concerned with the emissions of GHGs generated by human activity, including carbon dioxide (CO2), methane (CH4), nitrous oxide (N2O), tetrafluoromethane, hexafluoroethane, sulfur hexafluoride (SF6), HFC-23 (fluoroform), HFC-134a (1,1,1,2-tetrafluoroethane), and HFC-152a (difluoroethane). In the U.S., the main source of GHG emissions is electricity generation, followed by transportation. In California, transportation sources (including passenger cars, light-duty trucks, other trucks, buses, and motorcycles) make up the largest source of GHG-emitting sources. The dominant GHG emitted is CO2, mostly from fossil fuel combustion. -

Section 2. Hazards Identification OSHA/HCS Status : This Material Is Considered Hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200)
SAFETY DATA SHEET Nonflammable Gas Mixture: Ethane / Hexafluoroethane (R116) / Methyl Fluoride / Nitrogen / Nitrogen Trifluoride / Octafluorocyclobutane (C318) / Trifluoromethane (R23) Section 1. Identification GHS product identifier : Nonflammable Gas Mixture: Ethane / Hexafluoroethane (R116) / Methyl Fluoride / Nitrogen / Nitrogen Trifluoride / Octafluorocyclobutane (C318) / Trifluoromethane (R23) Other means of : Not available. identification Product use : Synthetic/Analytical chemistry. SDS # : 019126 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 24-hour telephone : 1-866-734-3438 Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : GASES UNDER PRESSURE - Compressed gas substance or mixture GHS label elements Hazard pictograms : Signal word : Warning Hazard statements : Contains gas under pressure; may explode if heated. May displace oxygen and cause rapid suffocation. Precautionary statements General : Read and follow all Safety Data Sheets (SDS’S) before use. Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Close valve after each use and when empty. Use equipment rated for cylinder pressure. Do not open valve until connected to equipment prepared for use. Use a back flow preventative device in the piping. Use only equipment of compatible materials of construction. Prevention : Not applicable. Response : Not applicable. Storage : Protect from sunlight when ambient temperature exceeds 52°C/125°F. Store in a well- ventilated place. Disposal : Not applicable. Hazards not otherwise : In addition to any other important health or physical hazards, this product may displace classified oxygen and cause rapid suffocation. -

SAFETY DATA SHEET Halocarbon R-508B
SAFETY DATA SHEET Halocarbon R-508B Section 1. Identification GHS product identifier : Halocarbon R-508B Other means of : Not available. identification Product use : Synthetic/Analytical chemistry. SDS # : 006720 Supplier's details : Airgas USA, LLC and its affiliates 259 North Radnor-Chester Road Suite 100 Radnor, PA 19087-5283 1-610-687-5253 Emergency telephone : 1-866-734-3438 number (with hours of operation) Section 2. Hazards identification OSHA/HCS status : This material is considered hazardous by the OSHA Hazard Communication Standard (29 CFR 1910.1200). Classification of the : GASES UNDER PRESSURE - Liquefied gas substance or mixture GHS label elements Hazard pictograms : Signal word : Warning Hazard statements : Contains gas under pressure; may explode if heated. May cause frostbite. May displace oxygen and cause rapid suffocation. Precautionary statements General : Read and follow all Safety Data Sheets (SDS’S) before use. Read label before use. Keep out of reach of children. If medical advice is needed, have product container or label at hand. Close valve after each use and when empty. Use equipment rated for cylinder pressure. Do not open valve until connected to equipment prepared for use. Use a back flow preventative device in the piping. Use only equipment of compatible materials of construction. Always keep container in upright position. Prevention : Use and store only outdoors or in a well ventilated place. Response : Not applicable. Storage : Protect from sunlight. Protect from sunlight when ambient temperature exceeds 52°C/125°F. Store in a well-ventilated place. Disposal : Not applicable. Hazards not otherwise : Liquid can cause burns similar to frostbite. classified Date of issue/Date of revision : 10/31/2014.