Gorilla Genome Structural Variation Reveals Evolutionary Parallelisms with Chimpanzee
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

An Evolutionary Based Strategy for Predicting Rational Mutations in G Protein-Coupled Receptors
Ecology and Evolutionary Biology 2021; 6(3): 53-77 http://www.sciencepublishinggroup.com/j/eeb doi: 10.11648/j.eeb.20210603.11 ISSN: 2575-3789 (Print); ISSN: 2575-3762 (Online) An Evolutionary Based Strategy for Predicting Rational Mutations in G Protein-Coupled Receptors Miguel Angel Fuertes*, Carlos Alonso Department of Microbiology, Centre for Molecular Biology “Severo Ochoa”, Spanish National Research Council and Autonomous University, Madrid, Spain Email address: *Corresponding author To cite this article: Miguel Angel Fuertes, Carlos Alonso. An Evolutionary Based Strategy for Predicting Rational Mutations in G Protein-Coupled Receptors. Ecology and Evolutionary Biology. Vol. 6, No. 3, 2021, pp. 53-77. doi: 10.11648/j.eeb.20210603.11 Received: April 24, 2021; Accepted: May 11, 2021; Published: July 13, 2021 Abstract: Capturing conserved patterns in genes and proteins is important for inferring phenotype prediction and evolutionary analysis. The study is focused on the conserved patterns of the G protein-coupled receptors, an important superfamily of receptors. Olfactory receptors represent more than 2% of our genome and constitute the largest family of G protein-coupled receptors, a key class of drug targets. As no crystallographic structures are available, mechanistic studies rely on the use of molecular dynamic modelling combined with site-directed mutagenesis data. In this paper, we hypothesized that human-mouse orthologs coding for G protein-coupled receptors maintain, at speciation events, shared compositional structures independent, to some extent, of their percent identity as reveals a method based in the categorization of nucleotide triplets by their gross composition. The data support the consistency of the hypothesis, showing in ortholog G protein-coupled receptors the presence of emergent shared compositional structures preserved at speciation events. -

WO 2019/068007 Al Figure 2
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2019/068007 Al 04 April 2019 (04.04.2019) W 1P O PCT (51) International Patent Classification: (72) Inventors; and C12N 15/10 (2006.01) C07K 16/28 (2006.01) (71) Applicants: GROSS, Gideon [EVIL]; IE-1-5 Address C12N 5/10 (2006.0 1) C12Q 1/6809 (20 18.0 1) M.P. Korazim, 1292200 Moshav Almagor (IL). GIBSON, C07K 14/705 (2006.01) A61P 35/00 (2006.01) Will [US/US]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., C07K 14/725 (2006.01) P.O. Box 4044, 7403635 Ness Ziona (TL). DAHARY, Dvir [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (21) International Application Number: Box 4044, 7403635 Ness Ziona (IL). BEIMAN, Merav PCT/US2018/053583 [EilL]; c/o ImmPACT-Bio Ltd., 2 Ilian Ramon St., P.O. (22) International Filing Date: Box 4044, 7403635 Ness Ziona (E.). 28 September 2018 (28.09.2018) (74) Agent: MACDOUGALL, Christina, A. et al; Morgan, (25) Filing Language: English Lewis & Bockius LLP, One Market, Spear Tower, SanFran- cisco, CA 94105 (US). (26) Publication Language: English (81) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of national protection available): AE, AG, AL, AM, 62/564,454 28 September 2017 (28.09.2017) US AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, 62/649,429 28 March 2018 (28.03.2018) US CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, (71) Applicant: IMMP ACT-BIO LTD. -

Gorilla Genome Structural Variation Reveals Evolutionary Parallelisms with Chimpanzee
Downloaded from genome.cshlp.org on September 30, 2021 - Published by Cold Spring Harbor Laboratory Press Gorilla genome structural variation reveals evolutionary parallelisms with chimpanzee Mario Ventura1,2, Claudia R. Catacchio1,2, Can Alkan1,3, Tomas Marques-Bonet1,4, Saba Sajjadian1, Tina A. Graves5, Fereydoun Hormozdiari6, Arcadi Navarro4, Maika Malig1, Carl Baker1, Choli Lee1, Emily H. Turner1, Lin Chen1, Jeffrey M. Kidd1,7, Nicoletta Archidiacono2, Jay Shendure1, Richard K. Wilson5, and Evan E. Eichler1,3† 1 Department of Genome Sciences, University of Washington School of Medicine, Seattle, Washington 98195, USA 2 Department of Genetics and Microbiology, University of Bari, Bari 70126, Italy 3 Howard Hughes Medical Institute, Seattle, Washington 98195, USA 4 IBE, Institut de Biologia Evolutiva (UPF-CSIC), Universitat Pompeu Fabra, PRBB, Doctor Aiguader, 88, 08003 Barcelona, Catalonia, Spain 5 Washington University Genome Sequencing Center, School of Medicine, St. Louis, Missouri 63108, USA 6 School of Computing Science, Simon Fraser University, Burnaby, British Columbia V5A 1S6, Canada 7 Department of Genetics, Stanford University, Stanford, California 94305, USA †Corresponding author: Evan E. Eichler, Ph.D. University of Washington School of Medicine Howard Hughes Medical Institute 3720 15th Avenue NE, S413C Box 355065 Seattle, WA 98195-5065 Phone: (206) 543-9526 E-mail: [email protected] MV, [email protected] CRC, [email protected] CA, [email protected] TMB, [email protected] SS, [email protected] -

(19) United States (12) Patent Application Publication (10) Pub
US 20140004512A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2014/0004512 A1 Santourlidis (43) Pub. Date: Jan. 2, 2014 (54) QUALITY DETERMINATION OF STEM Publication Classi?cation CELLS (51) Int. Cl. (75) Inventor: Simeon Santourlidis, Neuss (DE) C12Q 1/68 (200601) , __ (52) US. Cl. (73) Asslgnees: Peter _W‘?r“e__t’ Dusseldorf (DE); F‘med CPC .................................. .. C12Q 1/6888 (2013.01) Ghanjatl, Dusseldorf (DE); Simeon USPC 435/6 11 Santourlidis, Neuss (DE); Nicole ....................................................... .. Groth, Grefrath (DE) (21) Appl. No.: 13/878,915 (57) ABSTRACT (22) PCT Filed: 06L 11, 2011 The invention relates to a method for determining the quality of a pluripotent stem cell, comprising the following steps: (86) PCT NOJ PCT/EP11/67697 measuring the DNA methylation of at least one CpG in a CpG §371 (OX1), island in at least tWo genes of the pluripotent stem cell, (2)’ (4) Date; sep_ 18, 2013 comparing it to the DNA methylation of the at least one CpG in a CpG island in the at least tWo genes of at least one ( 30 ) Forei g n APP lication Priorit y Data reference cell, Wherein the genes are located on different chromosomes and belong to the gene family of olfactory Oct. 11, 2010 (EP) ................................ .. 101871374 receptor genes. Patent Application Publication Jan. 2, 2014 Sheet 2 0f 6 US 2014/0004512 A1 B8595 mmmH ____A_a 216m321 .1 41.__~ wwwmmHmw 00000O00O RRRRRR Patent Application Publication Jan. 2, 2014 Sheet 5 0f 6 US 2014/0004512 A1 mmm —— 0R1E1 2.065051 . mHmEnoEE‘mm: OR3M OR3A1 ——. ORBAZ 0mm UR1A2 Patent Application Publication Jan. -

The Hypothalamus As a Hub for SARS-Cov-2 Brain Infection and Pathogenesis
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. The hypothalamus as a hub for SARS-CoV-2 brain infection and pathogenesis Sreekala Nampoothiri1,2#, Florent Sauve1,2#, Gaëtan Ternier1,2ƒ, Daniela Fernandois1,2 ƒ, Caio Coelho1,2, Monica ImBernon1,2, Eleonora Deligia1,2, Romain PerBet1, Vincent Florent1,2,3, Marc Baroncini1,2, Florence Pasquier1,4, François Trottein5, Claude-Alain Maurage1,2, Virginie Mattot1,2‡, Paolo GiacoBini1,2‡, S. Rasika1,2‡*, Vincent Prevot1,2‡* 1 Univ. Lille, Inserm, CHU Lille, Lille Neuroscience & Cognition, DistAlz, UMR-S 1172, Lille, France 2 LaBoratorY of Development and PlasticitY of the Neuroendocrine Brain, FHU 1000 daYs for health, EGID, School of Medicine, Lille, France 3 Nutrition, Arras General Hospital, Arras, France 4 Centre mémoire ressources et recherche, CHU Lille, LiCEND, Lille, France 5 Univ. Lille, CNRS, INSERM, CHU Lille, Institut Pasteur de Lille, U1019 - UMR 8204 - CIIL - Center for Infection and ImmunitY of Lille (CIIL), Lille, France. # and ƒ These authors contriButed equallY to this work. ‡ These authors directed this work *Correspondence to: [email protected] and [email protected] Short title: Covid-19: the hypothalamic hypothesis 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.06.08.139329; this version posted June 19, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Us 2018 / 0305689 A1
US 20180305689A1 ( 19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0305689 A1 Sætrom et al. ( 43 ) Pub . Date: Oct. 25 , 2018 ( 54 ) SARNA COMPOSITIONS AND METHODS OF plication No . 62 /150 , 895 , filed on Apr. 22 , 2015 , USE provisional application No . 62/ 150 ,904 , filed on Apr. 22 , 2015 , provisional application No. 62 / 150 , 908 , (71 ) Applicant: MINA THERAPEUTICS LIMITED , filed on Apr. 22 , 2015 , provisional application No. LONDON (GB ) 62 / 150 , 900 , filed on Apr. 22 , 2015 . (72 ) Inventors : Pål Sætrom , Trondheim (NO ) ; Endre Publication Classification Bakken Stovner , Trondheim (NO ) (51 ) Int . CI. C12N 15 / 113 (2006 .01 ) (21 ) Appl. No. : 15 /568 , 046 (52 ) U . S . CI. (22 ) PCT Filed : Apr. 21 , 2016 CPC .. .. .. C12N 15 / 113 ( 2013 .01 ) ; C12N 2310 / 34 ( 2013. 01 ) ; C12N 2310 /14 (2013 . 01 ) ; C12N ( 86 ) PCT No .: PCT/ GB2016 /051116 2310 / 11 (2013 .01 ) $ 371 ( c ) ( 1 ) , ( 2 ) Date : Oct . 20 , 2017 (57 ) ABSTRACT The invention relates to oligonucleotides , e . g . , saRNAS Related U . S . Application Data useful in upregulating the expression of a target gene and (60 ) Provisional application No . 62 / 150 ,892 , filed on Apr. therapeutic compositions comprising such oligonucleotides . 22 , 2015 , provisional application No . 62 / 150 ,893 , Methods of using the oligonucleotides and the therapeutic filed on Apr. 22 , 2015 , provisional application No . compositions are also provided . 62 / 150 ,897 , filed on Apr. 22 , 2015 , provisional ap Specification includes a Sequence Listing . SARNA sense strand (Fessenger 3 ' SARNA antisense strand (Guide ) Mathew, Si Target antisense RNA transcript, e . g . NAT Target Coding strand Gene Transcription start site ( T55 ) TY{ { ? ? Targeted Target transcript , e . -
Explorations in Olfactory Receptor Structure and Function by Jianghai
Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] Dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 ABSTRACT Explorations in Olfactory Receptor Structure and Function by Jianghai Ho Department of Neurobiology Duke University Date:_______________________ Approved: ___________________________ Hiroaki Matsunami, Supervisor ___________________________ Jorg Grandl, Chair ___________________________ Marc Caron ___________________________ Sid Simon ___________________________ [Committee Member Name] An abstract of a dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department of Neurobiology in the Graduate School of Duke University 2014 Copyright by Jianghai Ho 2014 Abstract Olfaction is one of the most primitive of our senses, and the olfactory receptors that mediate this very important chemical sense comprise the largest family of genes in the mammalian genome. It is therefore surprising that we understand so little of how olfactory receptors work. In particular we have a poor idea of what chemicals are detected by most of the olfactory receptors in the genome, and for those receptors which we have paired with ligands, we know relatively little about how the structure of these ligands can either activate or inhibit the activation of these receptors. Furthermore the large repertoire of olfactory receptors, which belong to the G protein coupled receptor (GPCR) superfamily, can serve as a model to contribute to our broader understanding of GPCR-ligand binding, especially since GPCRs are important pharmaceutical targets. -
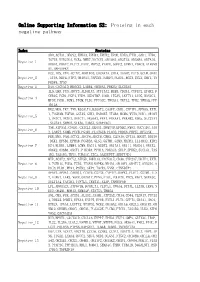
Online Supporting Information S2: Proteins in Each Negative Pathway
Online Supporting Information S2: Proteins in each negative pathway Index Proteins ADO,ACTA1,DEGS2,EPHA3,EPHB4,EPHX2,EPOR,EREG,FTH1,GAD1,HTR6, IGF1R,KIR2DL4,NCR3,NME7,NOTCH1,OR10S1,OR2T33,OR56B4,OR7A10, Negative_1 OR8G1,PDGFC,PLCZ1,PROC,PRPS2,PTAFR,SGPP2,STMN1,VDAC3,ATP6V0 A1,MAPKAPK2 DCC,IDS,VTN,ACTN2,AKR1B10,CACNA1A,CHIA,DAAM2,FUT5,GCLM,GNAZ Negative_2 ,ITPA,NEU4,NTF3,OR10A3,PAPSS1,PARD3,PLOD1,RGS3,SCLY,SHC1,TN FRSF4,TP53 Negative_3 DAO,CACNA1D,HMGCS2,LAMB4,OR56A3,PRKCQ,SLC25A5 IL5,LHB,PGD,ADCY3,ALDH1A3,ATP13A2,BUB3,CD244,CYFIP2,EPHX2,F CER1G,FGD1,FGF4,FZD9,HSD17B7,IL6R,ITGAV,LEFTY1,LIPG,MAN1C1, Negative_4 MPDZ,PGM1,PGM3,PIGM,PLD1,PPP3CC,TBXAS1,TKTL2,TPH2,YWHAQ,PPP 1R12A HK2,MOS,TKT,TNN,B3GALT4,B3GAT3,CASP7,CDH1,CYFIP1,EFNA5,EXTL 1,FCGR3B,FGF20,GSTA5,GUK1,HSD3B7,ITGB4,MCM6,MYH3,NOD1,OR10H Negative_5 1,OR1C1,OR1E1,OR4C11,OR56A3,PPA1,PRKAA1,PRKAB2,RDH5,SLC27A1 ,SLC2A4,SMPD2,STK36,THBS1,SERPINC1 TNR,ATP5A1,CNGB1,CX3CL1,DEGS1,DNMT3B,EFNB2,FMO2,GUCY1B3,JAG Negative_6 2,LARS2,NUMB,PCCB,PGAM1,PLA2G1B,PLOD2,PRDX6,PRPS1,RFXANK FER,MVD,PAH,ACTC1,ADCY4,ADCY8,CBR3,CLDN16,CPT1A,DDOST,DDX56 ,DKK1,EFNB1,EPHA8,FCGR3A,GLS2,GSTM1,GZMB,HADHA,IL13RA2,KIR2 Negative_7 DS4,KLRK1,LAMB4,LGMN,MAGI1,NUDT2,OR13A1,OR1I1,OR4D11,OR4X2, OR6K2,OR8B4,OXCT1,PIK3R4,PPM1A,PRKAG3,SELP,SPHK2,SUCLG1,TAS 1R2,TAS1R3,THY1,TUBA1C,ZIC2,AASDHPPT,SERPIND1 MTR,ACAT2,ADCY2,ATP5D,BMPR1A,CACNA1E,CD38,CYP2A7,DDIT4,EXTL Negative_8 1,FCER1G,FGD3,FZD5,ITGAM,MAPK8,NR4A1,OR10V1,OR4F17,OR52D1,O R8J3,PLD1,PPA1,PSEN2,SKP1,TACR3,VNN1,CTNNBIP1 APAF1,APOA1,CARD11,CCDC6,CSF3R,CYP4F2,DAPK1,FLOT1,GSTM1,IL2 -

Supplemental Table S18. Cellular Process Enrichment Analysis Output
Supplemental Table S18. -

-

Supporting Information SURVIVAL and DIVERGENCE in a SMALL GROUP: the EXTRAORDINARY GENOMIC HISTORY of the ENDANGERED APENNINE BROWN BEAR
Supporting Information SURVIVAL AND DIVERGENCE IN A SMALL GROUP: THE EXTRAORDINARY GENOMIC HISTORY OF THE ENDANGERED APENNINE BROWN BEAR Andrea Benazzoa,*, Emiliano Trucchia,b,*, James A. Cahillc, Pierpaolo Maisano Delserd,e,f, Stefano Monad,e, Matteo Fumagallig , Lynsey Bunnefeldh,i, Luca Cornettij, Silvia Ghirottoa, Matteo Girardik, Lino Omettol,m, Alex Panzieraa, Omar Rota-Stabellil, Enrico Zanettia, Alexandros Karamanlidisn, Claudio Groffo, Ladislav Paulep, Leonardo Gentileq, Carles Vilàr, Saverio Vicarios, Luigi Boitanit, Ludovic Orlandou, Silvia Fusellia, Cristiano Vernesik, Beth Shapiroc, Paolo Ciuccit, Giorgio Bertorellea,1. aDepartment of Life Sciences and Biotechnology, University of Ferrara, 44121 Ferrara, Italy bCentre for Ecological and Evolutionary Synthesis (CEES), Department of Biosciences, University of Oslo, Oslo 1066, Norway cDepartment of Ecology and Evolutionary Biology, University of California, Santa Cruz, CA 95064, USA dInstitute de Systematics, Evolution, Biodiversite, ISYEB-UMR 7205-CNRS, MNHN, UPMC, EPHE, Ecole Pratique de Hautes Etudes, CP39, 75005, Paris, France eEPHE, PSL Research University, 75005, Paris, France fSmurfit Institute of Genetics, University of Dublin, Trinity College, Dublin 2, Ireland. gDepartment of Life Sciences, Silwood Park Campus, Imperial College London, Ascot, SL5 7PY, United Kingdom hInstitute of Evolutionary Biology, University of Edinburgh, Edinburgh, EH9 3FL, United Kingdom iBiological and Environmental Science, University of Stirling, Stirling, FK9 4LA, United Kingdom jZoological -

Supplementary Methods
doi: 10.1038/nature06162 SUPPLEMENTARY INFORMATION Supplementary Methods Cloning of human odorant receptors 423 human odorant receptors were cloned with sequence information from The Olfactory Receptor Database (http://senselab.med.yale.edu/senselab/ORDB/default.asp). Of these, 335 were predicted to encode functional receptors, 45 were predicted to encode pseudogenes, 29 were putative variant pairs of the same genes, and 14 were duplicates. We adopted the nomenclature proposed by Doron Lancet 1. OR7D4 and the six intact odorant receptor genes in the OR7D4 gene cluster (OR1M1, OR7G2, OR7G1, OR7G3, OR7D2, and OR7E24) were used for functional analyses. SNPs in these odorant receptors were identified from the NCBI dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP) or through genotyping. OR7D4 single nucleotide variants were generated by cloning the reference sequence from a subject or by inducing polymorphic SNPs by site-directed mutagenesis using overlap extension PCR. Single nucleotide and frameshift variants for the six intact odorant receptors in the same gene cluster as OR7D4 were generated by cloning the respective genes from the genomic DNA of each subject. The chimpanzee OR7D4 orthologue was amplified from chimpanzee genomic DNA (Coriell Cell Repositories). Odorant receptors that contain the first 20 amino acids of human rhodopsin tag 2 in pCI (Promega) were expressed in the Hana3A cell line along with a short form of mRTP1 called RTP1S, (M37 to the C-terminal end), which enhances functional expression of the odorant receptors 3. For experiments with untagged odorant receptors, OR7D4 RT and S84N variants without the Rho tag were cloned into pCI.