The Genomic Revolution and Species Delimitation in Birds (And Other
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Malawi Trip Report 12Th to 28Th September 2014
Malawi Trip Report 12th to 28th September 2014 Bohm’s Bee-eater by Keith Valentine Trip Report compiled by Tour Leader: Keith Valentine RBT Malawi Trip Report September 2014 2 Top 10 Birds: 1. Scarlet-tufted Sunbird 2. Pel’s Fishing Owl 3. Lesser Seedcracker 4. Thyolo Alethe 5. White-winged Apalis 6. Racket-tailed Roller 7. Blue Swallow 8. Bohm’s Flycatcher 9. Babbling Starling 10. Bohm’s Bee-eater/Yellow-throated Apalis Top 5 Mammals: 1. African Civet 2. Four-toed Elephant Shrew 3. Sable Antelope 4. Bush Pig 5. Side-striped Jackal/Greater Galago/Roan Antelope/Blotched Genet Trip Summary This was our first ever fully comprehensive tour to Malawi and was quite simply a fantastic experience in all respects. For starters, many of the accommodations are of excellent quality and are also situated in prime birding locations with a large number of the area’s major birding targets found in close proximity. The food is generally very good and the stores and lodges are for the most part stocked with decent beer and a fair selection of South African wine. However, it is the habitat diversity that is largely what makes Malawi so good from a birding point of view. Even though it is a small country, this good variety of habitat, and infrastructure that allows access to these key zones, insures that the list of specials is long and attractive. Our tour was extremely successful in locating the vast majority of the region’s most wanted birds and highlights included Red-winged Francolin, White-backed Night Heron, African Cuckoo-Hawk, Western Banded Snake -

Henicorhina Anachoreta (Troglodytidae), Another Endemic Bird Species for the Sierra Nevada De Santa Marta, Colombia
Henicorhina anachoreta (Troglodytidae), another endemic bird Breve Nota species for the Sierra Nevada de Santa Marta, Colombia Henichorhina anachoreta (Troglodytidae), otra especie de ave endémica de la Sierra Nevada de Santa Marta, Colombia Carlos Daniel Cadena1, Lina María Caro1, Paula C. Caycedo2, Andrés M. Cuervo1,3, Rauri C. K. Bowie4 & Hans Slabbekoorn5 1Departamento de Ciencias Biológicas, Universidad de los Andes, Bogotá, Colombia. 2Institute of Biodiversity and Ecosystem Dynamics, Ecology and Evolution Program, Universiteit van Amsterdam, Amsterdam, Ornitología Colombiana Ornitología The Netherlands 3Department of Ecology & Evolutionary Biology, Tulane University, New Orleans, USA 4Museum of Vertebrate Zoology and Department of Integrative Biology, University of California, Berkeley, USA 5Behavioral Biology, Institute of Biology, Leiden University, Leiden, The Netherlands [email protected] Abstract In a previous study, we presented evidence that the Henicorhina wood-wren inhabiting the upper slopes of the Sierra Neva- da de Santa Marta, H. anachoreta, merits status as a species distinct from the lower élevation taxon, H. bangsi, based on genetic and phenotypic evidence as well as differences in song. Moreover, in a narrow zone of sympatry we found that they showed differential responses to the songs of their own vs. the other form. However, we did not present the differences in plumage and morphometrics in detail, and did not make a formal taxonomic recommendation regarding their taxonomic status. We do so here, and present more detailed description of the differences in plumage and morphometrics in support of this recommendation. colombiana/ colombiana/ - Key words: Henichorhina wrens, speciation, Sierra Nevada de Santa Marta, taxonomy. Resumen ornitologia - En un estudio previo, presentamos evidencia que el cucaracho del género Henichorhina que habita las elevaciones superio- res de la Sierra Nevada de Santa Marta, H. -

The Best of Costa Rica March 19–31, 2019
THE BEST OF COSTA RICA MARCH 19–31, 2019 Buffy-crowned Wood-Partridge © David Ascanio LEADERS: DAVID ASCANIO & MAURICIO CHINCHILLA LIST COMPILED BY: DAVID ASCANIO VICTOR EMANUEL NATURE TOURS, INC. 2525 WALLINGWOOD DRIVE, SUITE 1003 AUSTIN, TEXAS 78746 WWW.VENTBIRD.COM THE BEST OF COSTA RICA March 19–31, 2019 By David Ascanio Photo album: https://www.flickr.com/photos/davidascanio/albums/72157706650233041 It’s about 02:00 AM in San José, and we are listening to the widespread and ubiquitous Clay-colored Robin singing outside our hotel windows. Yet, it was still too early to experience the real explosion of bird song, which usually happens after dawn. Then, after 05:30 AM, the chorus started when a vocal Great Kiskadee broke the morning silence, followed by the scratchy notes of two Hoffmann´s Woodpeckers, a nesting pair of Inca Doves, the ascending and monotonous song of the Yellow-bellied Elaenia, and the cacophony of an (apparently!) engaged pair of Rufous-naped Wrens. This was indeed a warm welcome to magical Costa Rica! To complement the first morning of birding, two boreal migrants, Baltimore Orioles and a Tennessee Warbler, joined the bird feast just outside the hotel area. Broad-billed Motmot . Photo: D. Ascanio © Victor Emanuel Nature Tours 2 The Best of Costa Rica, 2019 After breakfast, we drove towards the volcanic ring of Costa Rica. Circling the slope of Poas volcano, we eventually reached the inspiring Bosque de Paz. With its hummingbird feeders and trails transecting a beautiful moss-covered forest, this lodge offered us the opportunity to see one of Costa Rica´s most difficult-to-see Grallaridae, the Scaled Antpitta. -

21 Sep 2018 Lists of Victims and Hosts of the Parasitic
version: 21 Sep 2018 Lists of victims and hosts of the parasitic cowbirds (Molothrus). Peter E. Lowther, Field Museum Brood parasitism is an awkward term to describe an interaction between two species in which, as in predator-prey relationships, one species gains at the expense of the other. Brood parasites "prey" upon parental care. Victimized species usually have reduced breeding success, partly because of the additional cost of caring for alien eggs and young, and partly because of the behavior of brood parasites (both adults and young) which may directly and adversely affect the survival of the victim's own eggs or young. About 1% of all bird species, among 7 families, are brood parasites. The 5 species of brood parasitic “cowbirds” are currently all treated as members of the genus Molothrus. Host selection is an active process. Not all species co-occurring with brood parasites are equally likely to be selected nor are they of equal quality as hosts. Rather, to varying degrees, brood parasites are specialized for certain categories of hosts. Brood parasites may rely on a single host species to rear their young or may distribute their eggs among many species, seemingly without regard to any characteristics of potential hosts. Lists of species are not the best means to describe interactions between a brood parasitic species and its hosts. Such lists do not necessarily reflect the taxonomy used by the brood parasites themselves nor do they accurately reflect the complex interactions within bird communities (see Ortega 1998: 183-184). Host lists do, however, offer some insight into the process of host selection and do emphasize the wide variety of features than can impact on host selection. -

Bird) Species List
Aves (Bird) Species List Higher Classification1 Kingdom: Animalia, Phyllum: Chordata, Class: Reptilia, Diapsida, Archosauria, Aves Order (O:) and Family (F:) English Name2 Scientific Name3 O: Tinamiformes (Tinamous) F: Tinamidae (Tinamous) Great Tinamou Tinamus major Highland Tinamou Nothocercus bonapartei O: Galliformes (Turkeys, Pheasants & Quail) F: Cracidae Black Guan Chamaepetes unicolor (Chachalacas, Guans & Curassows) Gray-headed Chachalaca Ortalis cinereiceps F: Odontophoridae (New World Quail) Black-breasted Wood-quail Odontophorus leucolaemus Buffy-crowned Wood-Partridge Dendrortyx leucophrys Marbled Wood-Quail Odontophorus gujanensis Spotted Wood-Quail Odontophorus guttatus O: Suliformes (Cormorants) F: Fregatidae (Frigatebirds) Magnificent Frigatebird Fregata magnificens O: Pelecaniformes (Pelicans, Tropicbirds & Allies) F: Ardeidae (Herons, Egrets & Bitterns) Cattle Egret Bubulcus ibis O: Charadriiformes (Sandpipers & Allies) F: Scolopacidae (Sandpipers) Spotted Sandpiper Actitis macularius O: Gruiformes (Cranes & Allies) F: Rallidae (Rails) Gray-Cowled Wood-Rail Aramides cajaneus O: Accipitriformes (Diurnal Birds of Prey) F: Cathartidae (Vultures & Condors) Black Vulture Coragyps atratus Turkey Vulture Cathartes aura F: Pandionidae (Osprey) Osprey Pandion haliaetus F: Accipitridae (Hawks, Eagles & Kites) Barred Hawk Morphnarchus princeps Broad-winged Hawk Buteo platypterus Double-toothed Kite Harpagus bidentatus Gray-headed Kite Leptodon cayanensis Northern Harrier Circus cyaneus Ornate Hawk-Eagle Spizaetus ornatus Red-tailed -

Sierra Leone
SIERRA LEONE 9 - 24 FEBRUARY 2008 TOUR REPORT LEADER: NIK BORROW Our first exploratory tour to Sierra Leone was pretty tough going at times but certainly pulled a few goodies out of the bag! A respectable total of 305 species were recorded of which all but 12 were seen. The notable major highlights had to be the wonderful views of the amazing Yellow-headed Picathartes preening and posing at their nest site before going to roost, the restricted range Turati’s Boubou and no less than four stunning Gola Malimbes for everyone! Singing Brown Nightjars were discovered, sublime Egyptian Plovers enjoyed, colourful Buff-throated Sunbirds enthralled and secretive Capuchin Babblers were tracked down. Mammals were sparse but we had great looks at the beautiful Diana Monkey and Olive Colobus and we even almost saw a Pygmy Hippo that crashed away from us through the undergrowth! Other specialties included Red-chested Goshawk, Latham’s Forest Francolin, Black-shouldered and Standard-winged Nightjars, Blue-headed Bee-eater, Brown- cheeked and Yellow-casqued Hornbills, Hairy-breasted Barbet, Spotted Honeyguide, Little Green, Melancholy and Fire-bellied Woodpeckers, Fanti Saw-wing, Preuss’s Cliff Swallow, Pied-winged Swallow, Green-tailed and Grey-headed Bristlebills, Western Bearded Greenbul, Yellow-bearded Greenbul, Western Forest Robin, White-tailed Alethe, Finsch’s Flycatcher Thrush, Forest Scrub Robin, Sharpe’s Apalis, Kemp’s Longbill, Olivaceous and Ussher’s Flycatchers, Red-cheeked Wattle-eye, Rufous-winged and Puvel’s Illadopsis, Red-billed Helmet-shrike, Copper-tailed Glossy and Emerald Starlings, Maxwell’s Black Weaver, Red-vented Malimbe, Yellow-winged Pytilia and Dybowski’s Twinspot. -

Miombo Ecoregion Vision Report
MIOMBO ECOREGION VISION REPORT Jonathan Timberlake & Emmanuel Chidumayo December 2001 (published 2011) Occasional Publications in Biodiversity No. 20 WWF - SARPO MIOMBO ECOREGION VISION REPORT 2001 (revised August 2011) by Jonathan Timberlake & Emmanuel Chidumayo Occasional Publications in Biodiversity No. 20 Biodiversity Foundation for Africa P.O. Box FM730, Famona, Bulawayo, Zimbabwe PREFACE The Miombo Ecoregion Vision Report was commissioned in 2001 by the Southern Africa Regional Programme Office of the World Wide Fund for Nature (WWF SARPO). It represented the culmination of an ecoregion reconnaissance process led by Bruce Byers (see Byers 2001a, 2001b), followed by an ecoregion-scale mapping process of taxa and areas of interest or importance for various ecological and bio-physical parameters. The report was then used as a basis for more detailed discussions during a series of national workshops held across the region in the early part of 2002. The main purpose of the reconnaissance and visioning process was to initially outline the bio-physical extent and properties of the so-called Miombo Ecoregion (in practice, a collection of smaller previously described ecoregions), to identify the main areas of potential conservation interest and to identify appropriate activities and areas for conservation action. The outline and some features of the Miombo Ecoregion (later termed the Miombo– Mopane Ecoregion by Conservation International, or the Miombo–Mopane Woodlands and Grasslands) are often mentioned (e.g. Burgess et al. 2004). However, apart from two booklets (WWF SARPO 2001, 2003), few details or justifications are publically available, although a modified outline can be found in Frost, Timberlake & Chidumayo (2002). Over the years numerous requests have been made to use and refer to the original document and maps, which had only very restricted distribution. -

Environmental Impact Assessment
ENVIRONMENTAL IMPACT ASSESSMENT For ARA MACAO Resort & Marina To be located in: Placencia, Stann Creek District Prepared by: Volume 1 January 2006 Table of Contents 1.0 Project Description & Layout Plan 1-1 1.01 Project Location and Description 1-1 1.02 The Physical Development Plans and the Description of the Facilities. 1-2 1.03 Plan Layout 1-5 1.04 Specifications for the Facilities and Forecast of Activities 1-7 1.05 Phases of Project Implementation 1-7 2.0 The Physical Environment 2-1 2.01 Topography 2-1 2.02 Climate 2-5 2.03 Geology 2-6 2.2 Project Facilities 2-10 3.0 Policy and Legal Administrative Framework 3-1 3.1 Policy 3-1 3.2 Legal Framework 3-2 3.3 Administrative Framework 3-4 3.4 The EIA Process 3-6 3.5 Permits and approvals required by the project 3-7 3.6 International And Regional Environmental Agreements 3-8 4.0 Flora and Fauna 4-1 4.1 Introduction 4-1 4.1.1 Mangrove Swamp 4-1 4.1.2 Freshwater Marsh and Swamps 4-1 4.1.3 Transitional Low Broadleaf Forest 4-1 4.2 Flora Survey 4-2 4.3 Avifaunal Survey 4-6 4.3.1 Results of bird census 4-6 4.4 Species of Key Conservation Concern 4-10 4.4.1 Reptiles 4-10 4.4.1.1 Crocodiles (Family Crocodylidae) 4-10 4.4.1.2 Spiny Tailed Iguana 4-11 4.4.1.3 Boa Constrictor 4-11 4.4.1.4 Mammals 4-11 4.5 Estimated Alteration of Vegetation 4-12 4.6 Impacts and Mitigation Measures 4-12 5.0 Water Resource 5-1 5.1 Occupancy Rate 5-1 5.2 Potable Water Demand 5-1 5.3 Water Source 5-3 5.3.1 Preferred Option 5-3 5.4 Water Supply Description 5-5 5.5 Ground and Surface Waters Analysis 5-5 5.5.1 Water Quality -

SPLITS, LUMPS and SHUFFLES Splits, Lumps and Shuffles Thomas S
>> SPLITS, LUMPS AND SHUFFLES Splits, lumps and shuffles Thomas S. Schulenberg Based on features including boot colour and tail shape, Booted Racket-tail Ocreatus underwoodii may be as many as four species. 1 ‘Anna’s Racket- tail’ O. (u.) annae, male, Cock-of-the-rock Lodge, 30 Neotropical Birding 22 Cuzco, Peru, August 2017 (Bradley Hacker). This series focuses on recent taxonomic proposals – descriptions of new taxa, splits, lumps or reorganisations – that are likely to be of greatest interest to birders. This latest instalment includes: new species of sabrewing, parrot (maybe), tapaculo, and yellow finch (perhaps); proposed splits in Booted Racket-tail, Russet Antshrike, White-backed Fire-eye (split city!), Collared Crescentchest, Olive-backed Foliage-gleaner, Musician Wren, Spotted Nightingale-Thrush, Yellowish and Short-billed Pipits, Black-and- rufous Warbling Finch, Pectoral and Saffron-billed Sparrows, and Unicolored Blackbird; a reassessment of an earlier proposed split in Black-billed Thrush; the (gasp!) possibility of the lump of South Georgia Pipit; and re- evaluations of two birds each known only from a single specimen. Racking up the racket-tails Venezuela to Bolivia; across its range, the puffy ‘boots’ (leg feathering) may be white or buffy, Booted Racket-tail Ocreatus underwoodii is one and the racket-tipped outer tail feathers may be of the most widespread, and one of the fanciest, straight, or so curved that the outermost rectrices hummingbirds of the Andes. It occurs from cross over one another. 2 ‘Peruvian’ Racket-tail O. (u.) peruanus, female, Abra Patricia, San Martín, Peru, October 2011 (Nick Athanas/antpitta.com). 3 ‘Peruvian Racket-tail’ O. -
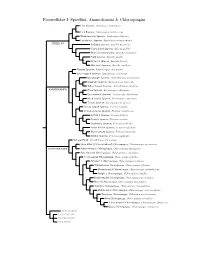
Passerellidae Species Tree
Passerellidae I: Spizellini, Ammodramini & Chlorospingini Lark Sparrow, Chondestes grammacus Lark Bunting, Calamospiza melanocorys Black-throated Sparrow, Amphispiza bilineata Five-striped Sparrow, Amphispiza quinquestriata SPIZELLINI Chipping Sparrow, Spizella passerina Clay-colored Sparrow, Spizella pallida Black-chinned Sparrow, Spizella atrogularis Field Sparrow, Spizella pusilla Brewer’s Sparrow, Spizella breweri Worthen’s Sparrow, Spizella wortheni Tumbes Sparrow, Rhynchospiza stolzmanni Stripe-capped Sparrow, Rhynchospiza strigiceps Grasshopper Sparrow, Ammodramus savannarum Grassland Sparrow, Ammodramus humeralis Yellow-browed Sparrow, Ammodramus aurifrons AMMODRAMINI Olive Sparrow, Arremonops rufivirgatus Green-backed Sparrow, Arremonops chloronotus Black-striped Sparrow, Arremonops conirostris Tocuyo Sparrow, Arremonops tocuyensis Rufous-winged Sparrow, Peucaea carpalis Cinnamon-tailed Sparrow, Peucaea sumichrasti Botteri’s Sparrow, Peucaea botterii Cassin’s Sparrow, Peucaea cassinii Bachman’s Sparrow, Peucaea aestivalis Stripe-headed Sparrow, Peucaea ruficauda Black-chested Sparrow, Peucaea humeralis Bridled Sparrow, Peucaea mystacalis Tanager Finch, Oreothraupis arremonops Short-billed (Yellow-whiskered) Chlorospingus, Chlorospingus parvirostris CHLOROSPINGINI Yellow-throated Chlorospingus, Chlorospingus flavigularis Ashy-throated Chlorospingus, Chlorospingus canigularis Sooty-capped Chlorospingus, Chlorospingus pileatus Wetmore’s Chlorospingus, Chlorospingus wetmorei White-fronted Chlorospingus, Chlorospingus albifrons Brown-headed -

The Parelius Bird Collection from Ivory Coast at the Field Museum of Natural History, and the First Country Record of Rufous Cisticola Cisticola Rufus
2014 Notes Courtes 119 casqués fuient quand ils sont approchés de moins d’une vingtaine de mètres, et les guides du Parc national nous ont dit qu’ils n’avaient jamais auparavant vu ce comportement. Peut-être les oiseaux suivaient-ils un chemin habituel. Nous remercions Lamin Sanyang, conservateur du Parc National de Niumi, qui nous a accompagnés lors de notre visite en Gambie, et Jean-Louis Faure, membre du Groupe Ornithologique Normand, pour sa contribution à la validation des observations. Bibliographie BORROW, N. & DEMEY, R. (2011) Birds of Senegal and The Gambia. Christopher Helm, Londres. GÉROUDET, P. (1994), Grands Échassiers, Gallinacées, Râles d’Europe. Delachaux et Niestlé, Lausanne. LE NEVÉ, A., BARGAIN, B., TEGETMEYER, C. & GUYOT, G. (2013) Hivernage au Sénégal d’un Phragmite aquatique Acrocephalus paludicola “isabelle”: longévité de l’espèce et liens de migration. Malimbus 35: 142–146. Reçu 31 mai 2014 Revu 11 août 2014 1 2 3 Moussa Séga DIOP , Bertrand MENNESSON & John ROSE 1Cité SICA de Mbao Villa N° 191, BP 20077 Dakar-Thiaroye, Sénégal; <[email protected]> 2Co-coordinateur d’ornithologie, ANY, Villa de Chèvreloup, 34 route de Versailles, 78150 Rocquencourt, France 31 Bis rue des Châtre-Sacs, 92310 Sèvres, France The Parelius bird collection from Ivory Coast at the Field Museum of Natural History, and the first country record of Rufous Cisticola Cisticola rufus Museum collections can play a critical role in unravelling a country’s faunistic history. Each specimen creates a scientific, historical record of where and when a bird occurred, from which, among other things, we can learn about changes in land cover and climate, and infer changes in an organism’s distribution over time (e.g. -

Remote Tanzania
The wonderful Usambara Eagle Owl... our inevitable bird of the trip! (Pete Morris) REMOTE TANZANIA 27 SEPTEMBER / 1 – 16 / 24 OCTOBER 2016 LEADER: PETE MORRIS Our 2017 tour to ‘Remote Tanzania’ turned out to be a fantastic adventure that way-surpassed most of our expectations. And with the addition of a pre-tour Northern Tanzania endemics extension, and a post-tour Ud- zungwa Mountains extension, the tour really was converted to an ‘Ultimate Tanzania’. For most of the tour we were accompanied by our ever reliable, super friendly and effcient local driver and excellent local guide, and were blessed with largely good weather, most importantly when we were camping up in the mountains. With such a good set up and conditions, there were few excuses, so we effciently set about our task of hoovering up the endemics, whilst enjoying the other wonders that this great country has to offer. 1 BirdQuest Tour Report: Remote Tanzania 2016 www.birdquest-tours.com The stunning Swynnerton’s Robin... a highlight of the Udzungwa Extension (Pete Morris) The pre-tour extension involved a whistle stop trek from Arusha to the Lariboro Plains and then down to Ndutu on the edge of the great Serengeti Plains and back via the amazing Ngorongoro Crater. Our task was to fnd the endemics: Grey-breasted Spurfowl, Yellow-collared and Fischer’s Lovebirds, Beesley’s Lark, Rufous-tailed Weaver and Ashy Starling all performed well alongside an excellent cast of other hoped-for birds and mam- mals. The main tour saw us travelling through arid plains and on to the South Pare Mountains, the East and West Usambaras, the attractive Pemba Island, the remote Uluguru and Ukaguru Mountains, the impressive Mikumi National Park and its surrounding miombo woodlands and the little explored Kilombero Plains.