XRCC2 and XRCC3 Gene Polymorphism and Risk of Pancreatic Cancer
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

1 AGING Supplementary Table 2
SUPPLEMENTARY TABLES Supplementary Table 1. Details of the eight domain chains of KIAA0101. Serial IDENTITY MAX IN COMP- INTERFACE ID POSITION RESOLUTION EXPERIMENT TYPE number START STOP SCORE IDENTITY LEX WITH CAVITY A 4D2G_D 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ B 4D2G_E 52 - 69 52 69 100 100 2.65 Å PCNA X-RAY DIFFRACTION √ C 6EHT_D 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ D 6EHT_E 52 - 71 52 71 100 100 3.2Å PCNA X-RAY DIFFRACTION √ E 6GWS_D 41-72 41 72 100 100 3.2Å PCNA X-RAY DIFFRACTION √ F 6GWS_E 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ G 6GWS_F 41-72 41 72 100 100 2.9Å PCNA X-RAY DIFFRACTION √ H 6IIW_B 2-11 2 11 100 100 1.699Å UHRF1 X-RAY DIFFRACTION √ www.aging-us.com 1 AGING Supplementary Table 2. Significantly enriched gene ontology (GO) annotations (cellular components) of KIAA0101 in lung adenocarcinoma (LinkedOmics). Leading Description FDR Leading Edge Gene EdgeNum RAD51, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, CENPW, HJURP, NDC80, CDCA5, NCAPH, BUB1, ZWILCH, CENPK, KIF2C, AURKA, CENPN, TOP2A, CENPM, PLK1, ERCC6L, CDT1, CHEK1, SPAG5, CENPH, condensed 66 0 SPC24, NUP37, BLM, CENPE, BUB3, CDK2, FANCD2, CENPO, CENPF, BRCA1, DSN1, chromosome MKI67, NCAPG2, H2AFX, HMGB2, SUV39H1, CBX3, TUBG1, KNTC1, PPP1CC, SMC2, BANF1, NCAPD2, SKA2, NUP107, BRCA2, NUP85, ITGB3BP, SYCE2, TOPBP1, DMC1, SMC4, INCENP. RAD51, OIP5, CDK1, SPC25, CCNB1, BIRC5, NCAPG, ZWINT, MAD2L1, SKA3, NUF2, BUB1B, CENPA, SKA1, AURKB, NEK2, ESCO2, CENPW, HJURP, TTK, NDC80, CDCA5, BUB1, ZWILCH, CENPK, KIF2C, AURKA, DSCC1, CENPN, CDCA8, CENPM, PLK1, MCM6, ERCC6L, CDT1, HELLS, CHEK1, SPAG5, CENPH, PCNA, SPC24, CENPI, NUP37, FEN1, chromosomal 94 0 CENPL, BLM, KIF18A, CENPE, MCM4, BUB3, SUV39H2, MCM2, CDK2, PIF1, DNA2, region CENPO, CENPF, CHEK2, DSN1, H2AFX, MCM7, SUV39H1, MTBP, CBX3, RECQL4, KNTC1, PPP1CC, CENPP, CENPQ, PTGES3, NCAPD2, DYNLL1, SKA2, HAT1, NUP107, MCM5, MCM3, MSH2, BRCA2, NUP85, SSB, ITGB3BP, DMC1, INCENP, THOC3, XPO1, APEX1, XRCC5, KIF22, DCLRE1A, SEH1L, XRCC3, NSMCE2, RAD21. -

Rad51c Deficiency Destabilizes XRCC3, Impairs Recombination and Radiosensitizes S/G2-Phase Cells
Lawrence Berkeley National Laboratory Lawrence Berkeley National Laboratory Title Rad51C deficiency destabilizes XRCC3, impairs recombination and radiosensitizes S/G2- phase cells Permalink https://escholarship.org/uc/item/2fp0538b Authors Lio, Yi-Ching Schild, David Brenneman, Mark A. et al. Publication Date 2004-05-01 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Rad51C deficiency destabilizes XRCC3, impairs recombination and radiosensitizes S/G2-phase cells Yi-Ching Lio1, 2,*, David Schild1, Mark A. Brenneman3, J. Leslie Redpath2 and David J. Chen1 1Life Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA; 2Department of Radiation Oncology, University of California, Irvine, Irvine, CA 92697, USA; 3Department of Genetics, Rutgers University, Piscataway, NJ 08854, USA. * To whom correspondence should be addressed: Yi-Ching Lio MS74-157, Life Sciences Division Lawrence Berkeley National Laboratory One Cyclotron Road Berkeley, CA 94720 Phone: (510) 486-5861 Fax: (510) 486-6816 e-mail: [email protected] Running title: Human Rad51C functions in homologous recombination Total character count: 52621 1 ABSTRACT The highly conserved Rad51 protein plays an essential role in repairing DNA damage through homologous recombination. In vertebrates, five Rad51 paralogs (Rad51B, Rad51C, Rad51D, XRCC2, XRCC3) are expressed in mitotically growing cells, and are thought to play mediating roles in homologous recombination, though their precise functions remain unclear. Here we report the use of RNA interference to deplete expression of Rad51C protein in human HT1080 and HeLa cells. In HT1080 cells, depletion of Rad51C by small interfering RNA caused a significant reduction of frequency in homologous recombination. The level of XRCC3 protein was also sharply reduced in Rad51C-depleted HeLa cells, suggesting that XRCC3 is dependent for its stability upon heterodimerization with Rad51C. -

Contribution of DNA Double-Strand Break Repair Gene XRCC3 Genotypes to Oral Cancer Susceptibility in Taiwan
ANTICANCER RESEARCH 34: 2951-2956 (2014) Contribution of DNA Double-strand Break Repair Gene XRCC3 Genotypes to Oral Cancer Susceptibility in Taiwan CHIA-WEN TSAI1,3, WEN-SHIN CHANG2,3, JUHN-CHERNG LIU2,3, MING-HSUI TSAI3, CHENG-CHIEH LIN3,4 and DA-TIAN BAU1,2,3 Graduate Institutes of 1Basic Medical Science and 2Clinic Medical Science, China Medical University, Taichung, Taiwan, R.O.C.; 3Terry Fox Cancer Research Laboratory, Department of Medical Research, in China Medical University Hospital, Taichung, Taiwan, R.O.C.; 4Asia University, Taichung, Taiwan, R.O.C. Abstract. The DNA repair gene X-ray repair cross and neck cancers in Taiwan (2). Three major lifestyle factors, complementing protein 3 (XRCC3) is thought to play a major namely the use of tobacco, alcohol and betel nuts, are main role in double-strand break repair and in maintaining causes of oral cancer in Taiwan, while the genomic etiology genomic stability. Very possibly, defective double-strand of oral cancer is of great interest but largely unknown. break repair of cells can lead to carcinogenesis. Therefore, a Human DNA repair mechanisms protect the genome from case–control study was performed to reveal the contribution various insults caused by endogenous and exogenous DNA- of XRCC3 genotypes to individual oral cancer susceptibility. damaging agents (3) and defects in the DNA repair system In this hospital-based research, the association of XRCC3 are thought to be essential in tumorigenesis (4, 5). Therefore, rs1799794, rs45603942, rs861530, rs3212057, rs1799796, it is logical to suspect that some genetic variants of DNA rs861539, rs28903081 genotypes with oral cancer risk in a repair genes might contribute to oral cancer pathogenesis. -

(EZH2) Genotypes with Bladder Cancer Risk in Taiwan
ANTICANCER RESEARCH 36 : 4509-4514 (2016) doi:10.21873/anticanres.10997 Association of Enhancer of Zeste 2 ( EZH2 ) Genotypes with Bladder Cancer Risk in Taiwan WEN-SHIN CHANG 1,2* , CHENG-HSI LIAO 1,2,3,8* , CHIA-WEN TSAI 2* , PEI-SHIN HU 2,4 , HSI-CHIN WU 2, SHIH-WEI HSU 1,2,3,8 , CHIEH-LUN HSIAO 1,2 , CHANG-HSIEN HSU 5, YI-WEN HUNG 6 and DA-TIAN BAU 1,2,7 1Graduate Institute of Clinical Medical Science, China Medical University, Taichung, Taiwan, R.O.C.; 2Terry Fox Cancer Research Laboratory, China Medical University Hospital, Taichung, Taiwan, R.O.C.; 3Taichung Armed Forces General Hospital, Taichung, Taiwan, R.O.C.; 4Department of Ophthalmology, Changhua Christian Hospital, Changhua, Taiwan, R.O.C.; Departments of 5Business Administration, and 7Bioinformatics and Medical Engineering, Asia University, Taichung, Taiwan, R.O.C.; 6Department of Medicine Research, Taichung Veterans General Hospital, Taichung, Taiwan, R.O.C.; 8National Defence Medical Center, Taipei, Taiwan, R.O.C. Abstract. Aim: Bladder cancer is the sixth most common associated with the lower risk of bladder cancer development, cancer worldwide and its incidence is particularly high in especially among non-smokers. many developed regions including southwestern Taiwan. However, the genetic contribution to the etiology of bladder Bladder cancer is the most serious urinary neoplasm cancer is not well-understood. The aim of this study was to worldwide, with high incidence rates of approximately evaluate the association of the enhancer of zeste homolog 2 10/100,000 and 3/100,000 in more and less developed (EZH2) genotypes with Taiwan bladder cancer risk. -

RAD51C-Deficient Cancer Cells Are Highly Sensitive to the PARP Inhibitor Olaparib
Published OnlineFirst March 19, 2013; DOI: 10.1158/1535-7163.MCT-12-0950 Molecular Cancer Chemical Therapeutics Therapeutics RAD51C-Deficient Cancer Cells Are Highly Sensitive to the PARP Inhibitor Olaparib Ahrum Min1, Seock-Ah Im1,2, Young-Kwang Yoon1, Sang-Hyun Song1, Hyun-Jin Nam1, Hyung-Seok Hur1, Hwang-Phill Kim1, Kyung-Hun Lee1,2, Sae-Won Han1,2, Do-Youn Oh1,2, Tae-You Kim1,2,4, Mark J. O'Connor5, Woo-Ho Kim1,3, and Yung-Jue Bang1,2 Abstract A PARP inhibitor is a rationally designed targeted therapy for cancers with impaired DNA repair abilities. RAD51C is a paralog of RAD51 that has an important role in the DNA damage response. We found that cell lines sensitive to a novel oral PARP inhibitor, olaparib, had low levels of RAD51C expression using microarray analysis, and we therefore hypothesized that low expression of RAD51C may hamper the DNA repair process, resulting in increased sensitivity to olaparib. Compared with the cells with normal RAD51C expression levels, RAD51C-deficient cancer cells were more sensitive to olaparib, and a higher proportion underwent cell death by inducing G2–M cell-cycle arrest and apoptosis. The restoration of RAD51C in a sensitive cell line caused attenuation of olaparib sensitivity. In contrast, silencingofRAD51Cinaresistant cell line enhanced the sensitivity to olaparib, and the number of RAD51 foci decreased with ablated RAD51C expression. We also found the expression of RAD51C was downregulated in cancer cells due to epigenetic changes and RAD51C expression was low in some gastric cancer tissues. Furthermore, olaparib significantly suppressed RAD51C-deficient tumor growth in a xenograft model. -
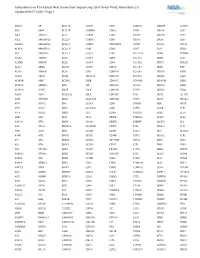
Comprehensive Pan-Cancer Next Generation Sequencing Solid Tumor Panel, Aberration List Updated 08-07-2020 --Page 1
Comprehensive Pan-Cancer Next Generation Sequencing Solid Tumor Panel, Aberration List Updated 08-07-2020 --Page 1 ABCC3 AR BCL11A CANT1 CDK1 CMKLR1 DAB2IP DUSP9 ABI1 ARAF BCL11B CAPRIN1 CDK12 CNBP DACH1 E2F1 ABL1 ARFRP1 BCL2 CAPZB CDK2 CNOT2 DACH2 E2F3 ABL2 ARHGAP20 BCL2A1 CARD11 CDK4 CNTN1 DAXX EBF1 ABLIM1 ARHGAP26 BCL2L1 CARM1 CDK5RAP2 CNTRL DCLK2 ECT2L ACACA ARHGEF12 BCL2L11 CARS CDK6 COG5 DCN EDIL3 ACE ARHGEF7 BCL2L2 CASC5 CDK7 COL11A1 DDB1 EDNRB ACER1 ARID1A BCL3 CASP3 CDK8 COL1A1 DDB2 EED ACSBG1 ARID1B BCL6 CASP7 CDK9 COL1A2 DDIT3 EEFSEC ACSL3 ARID2 BCL7A CASP8 CDKL5 COL3A1 DDR2 EGF ACSL6 ARID5B BCL9 CAV1 CDKN1A COL6A3 DDX10 EGFR ACVR1 ARIH2 BCOR CBFA2T3 CDKN1B COL9A3 DDX20 EGR1 ACVR1B ARNT BCORL1 CBFB CDKN1C COMMD1 DDX39B EGR2 ACVR1C ARRDC4 BCR CBL CDKN2A COX6C DDX3X EGR3 ACVR2A ASMTL BDNF CBLB CDKN2B CPNE1 DDX41 EGR4 ADD3 ASPH BHLHE22 CBLC CDKN2C CPS1 DDX5 EIF1AX ADM ASPSCR1 BICC1 CCDC28A CDKN2D CPSF6 DDX6 EIF4A2 AFF1 ASTN2 BIN1 CCDC6 CDX1 CRADD DEK EIF4E AFF3 ASXL1 BIRC3 CCDC88C CDX2 CREB1 DGKB ELF3 AFF4 ASXL2 BIRC6 CCK CEBPA CREB3L1 DGKI ELF4 AGR3 ATF1 BLM CCL2 CEBPB CREB3L2 DGKZ ELK4 AHCYL1 ATF3 BMP4 CCNA2 CEBPD CREBBP DICER1 ELL AHI1 ATG13 BMPR1A CCNB1IP1 CEBPE CRKL DIRAS3 ELN AHR ATG5 BRAF CCNB3 CENPF CRLF2 DIS3 ELOVL2 AHRR ATIC BRCA1 CCND1 CENPU CRTC1 DIS3L2 ELP2 AIP ATL1 BRCA2 CCND2 CEP170B CRTC3 DKK1 EML1 AK2 ATM BRCC3 CCND3 CEP57 CSF1 DKK2 EML4 AK5 ATP1B4 BRD1 CCNE1 CEP85L CSF1R DKK4 ENPP2 AKAP12 ATP8A2 BRD3 CCNG1 CHCHD7 CSF3 DLEC1 EP300 AKAP6 ATR BRD4 CCT6B CHD2 CSF3R DLL1 EP400 AKAP9 ATRNL1 BRIP1 CD19 CHD4 CSNK1A1 DLL3 -

132608682.Pdf
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Plymouth Electronic Archive and Research Library THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 278, No. 48, Issue of November 28, pp. 48357–48366, 2003 © 2003 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A. Functional Interaction between the Bloom’s Syndrome Helicase and the RAD51 Paralog, RAD51L3 (RAD51D)* Received for publication, August 11, 2003, and in revised form, September 12, 2003 Published, JBC Papers in Press, September 15, 2003, DOI 10.1074/jbc.M308838200 Jeremy P. Braybrooke‡§, Ji-Liang Li‡§, Leonard Wu‡, Fiona Caple¶, Fiona E. Benson¶, and Ian D. Hickson‡ʈ From ‡Cancer Research UK, Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford OX3 9DS, United Kingdom and the ¶Department of Biological Sciences, Lancaster Environment Centre, Lancaster University, Lancaster LA1 4YQ, United Kingdom Bloom’s syndrome (BS) is a genetic disorder associ- and plating, and a strikingly high level of chromosomal insta- ated with short stature, fertility defects, and a predispo- bility (3, 4). The characteristic feature of BS cells, which is used Downloaded from sition to the development of cancer. BS cells are char- in diagnosis of the disorder, is an elevated frequency of genetic acterized by genomic instability; in particular, a high recombination events, particularly sister-chromatid exchanges rate of reciprocal exchanges between sister-chromatids (SCEs) (5). However, this hyper-recombination is not limited to and homologous chromosomes. The BS gene product, exchanges between sister-chromatids, because interchromo- BLM, is a helicase belonging to the highly conserved somal homologous recombination also occurs at an elevated RecQ family. -

Transcriptome Profiling of Human Oocytes Experiencing Recurrent Total
www.nature.com/scientificreports OPEN Transcriptome profling of human oocytes experiencing recurrent total fertilization failure Received: 11 July 2018 Lun Suo1, Yu xiao Zhou2, Li ling Jia2, Hai bo Wu1, Jin Zheng2, Qi feng Lyu1, Li hua Sun1, Accepted: 16 November 2018 Han Sun3 & Yan ping Kuang1 Published: xx xx xxxx There exist some patients who face recurrent total fertilization failure during assisted reproduction treatment, but the pathological mechanism underlying is elusive. Here, by using sc-RNA-seq method, the transcriptome profles of ten abnormally fertilized zygotes were assessed, including fve zygotes from one patient with recurrent Poly-PN zygotes, and fve zygotes from a patient with pronuclear fusion failure. Four zygotes with three pronuclear (Tri-PN) were collected from four diferent patients as controls. After that, we identifed 951 and 1697 signifcantly diferentially expressed genes (SDEGs) in Poly-PN and PN arrest zygotes, respectively as compared with the control group. KEGG analyses indicated down regulated genes in the Poly-PN group included oocyte meiosis related genes, such as PPP2R1B, YWHAZ, MAD2L1, SPDYC, SKP1 and CDC27, together with genes associated with RNA processing, such as SF3B1, LOC645691, MAGOHB, PHF5A, PRPF18, DDX5, THOC1 and BAT1. In contrast, down regulated genes in the PN arrest group, included cell cycle genes, such as E2F4, DBF4, YWHAB, SKP2, CDC23, SMC3, CDC25A, CCND3, BUB1B, MDM2, CCNA2 and CDC7, together with homologous recombination related genes, such as NBN, XRCC3, SHFM1, RAD54B and RAD51. Thus, our work provides a better understanding of transcriptome profles underlying RTFF, although it based on a limited number of patients. Despite nearly forty years of scientifc and clinical advance in the feld of assisted reproduction, there still exist some rarely patients, even though rarely occur, who have to face recurrent total fertilization failure (RTFF) with- out any visual precautionary indicator1–3, even some of them could be rescued by assisted oocyte activation4. -

Frontgraphics Pt1r
Technology Transfer ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ○ ...................... .... Converting scientific knowledge into ransferring technology to equipment, or other resources. Funds the private sector, a pri- must come from the nongovernmental commercially useful mary mission of DOE, is partner. A benefit to participating com- products strongly encouraged in the panies is the opportunity to negotiate T Human Genome Program exclusive licenses for inventions arising ......................... to enhance the nation’s investment in from these collaborations. For periods research and technological competitive- through 1996, the CRADAs in place in ness. Human genome centers at the DOE Human Genome Program in- Lawrence Berkeley National Laboratory cluded the following: (LBNL), Lawrence Livermore National Laboratory (LLNL), and Los Alamos • LLNL with Applied Biosystems National Laboratory (LANL) provide Division of Perkin-Elmer Corporation opportunities for private companies to to develop analytical instrumentation collaborate on joint projects or use labo- for faster DNA sequencing instru- ratory resources. These opportunities in- mentation; clude access to information (including • LANL with Amgen, Inc., to develop databases), personnel, and special facili- bioassays for cell growth factors; ties; informal research collaborations; Cooperative Research and Development • Oak Ridge National Laboratory Agreements (CRADAs); and patent and (ORNL) with Darwin Molecular, -

Association of XRCC3 and XRCC4 Gene Polymorphisms, Family History
Journal of Human Genetics (2013) 58, 679–685 & 2013 The Japan Society of Human Genetics All rights reserved 1434-5161/13 www.nature.com/jhg ORIGINAL ARTICLE Association of XRCC3 and XRCC4 gene polymorphisms, family history of cancer and tobacco smoking with non-small-cell lung cancer in a Chinese population: a case–control study Fei He1, Shen-Chih Chang2, Gina Maria Wallar2, Zuo-Feng Zhang2 and Lin Cai1 Single-nucleotide polymorphisms (SNPs) of DNA repair genes have been reported to modify cancer risk. This study aimed to determine SNPs of the DNA repair genes X-ray repair cross-complementing group 3 (XRCC3) and X-ray cross-complementing group 4 (XRCC4) and their association with non-small-cell lung cancer (NSCLC) susceptibility in a Chinese population. A total of 507 NSCLC patients and 662 healthy controls were recruited for genotyping. Epidemiological and clinical data were also collected for association studies. The data showed that the rs1799794 G allele in the XRCC3 gene and minor allele carriers of XRCC4, including rs1056503 and rs9293337, were inversely associated with NSCLC risk (GG vs homozygote AA), whereas the rs861537 AG or AA genotype and XRCC4 rs6869366 had a significantly increased NSCLC risk. Furthermore, tobacco smoking over 26 pack-years, a family history of lung cancer, exposure to environmental tobacco smoke (ETS) and negative mental status were risk factors for developing NSCLC. This study suggests that SNPs of XRCC3 and XRCC4 and other environmental factors are risk factors for developing NSCLC in this Chinese Han population. Journal of Human Genetics (2013) 58, 679–685; doi:10.1038/jhg.2013.78; published online 8 August 2013 Keywords: case–control study; non-small-cell lung cancer; single-nucleotide polymorphism; XRCC3 and XRCC4 INTRODUCTION homologous recombination and non-homologous end-joining DNA Lung cancer was the most commonly diagnosed cancer and the repairs. -

RAD51, XRCC3, and XRCC2 Mutation Screening in Finnish
Pelttari et al. SpringerPlus (2015) 4:92 DOI 10.1186/s40064-015-0880-3 a SpringerOpen Journal RESEARCH Open Access RAD51, XRCC3, and XRCC2 mutation screening in Finnish breast cancer families Liisa M Pelttari1,JohannaIKiiski1, Salla Ranta1, Sara Vilske1,CarlBlomqvist2, Kristiina Aittomäki3 and Heli Nevanlinna1* Abstract Majority of the known breast cancer susceptibility genes have a role in DNA repair and the most important high-risk genes BRCA1 and BRCA2 are specifically involved in the homologous recombination repair (HRR) of DNA double-strand breaks. A central player in HRR is RAD51 that binds DNA at the damage site. The RAD51 paralogs RAD51B, RAD51C, RAD51D, XRCC2, and XRCC3 facilitate the binding of RAD51 to DNA. While germline mutations in RAD51C and RAD51D are associated with high ovarian cancer risk and RAD51B polymorphisms with breast cancer, the contribution of RAD51, XRCC3, and XRCC2 is more unclear. To investigate the role of RAD51, XRCC3, and XRCC2 in breast cancer predisposition and to identify putative recurrent founder mutations in the Finnish population where such mutations have been observed in most of the currently known susceptibility genes, we screened 182 familial Finnish breast or ovarian cancer patients for germline variation in the RAD51and XRCC3 genes and 342 patients for variation in XRCC2, with a subset of the patients selected on the basis of decreased RAD51 protein expression on tumors. We also performed haplotype analyses for 1516 breast cancer cases and 1234 controls to assess the common variation in these genes. No pathogenic mutations were detected in any of the genes and the distribution of haplotypes was similar between cases and controls.