Electromyography and Muscle Biopsy in Paediatric Neuromuscular Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Acute Limb Ischemia Secondary to Myositis- Induced Compartment Syndrome in a Patient with Human Immunodeficiency Virus Infection
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector Acute limb ischemia secondary to myositis- induced compartment syndrome in a patient with human immunodeficiency virus infection Russell Lam, MD, Peter H. Lin, MD, Suresh Alankar, MD, Qizhi Yao, MD, PhD, Ruth L. Bush, MD, Changyi Chen, MD, PhD, and Alan B. Lumsden, MD, Houston, Tex Myositis, while uncommon, develops more frequently in patients with human immunodeficiency virus infection. We report a case of acute lower leg ischemia caused by myositis in such a patient. Urgent four-compartment fasciotomy of the lower leg was performed, which decompressed the compartmental hypertension and reversed the arterial ischemia. This case underscores the importance of recognizing compartment syndrome as a cause of acute limb ischemia. (J Vasc Surg 2003;37:1103-5.) Compartment syndrome results from elevated pressure compartment was firm and tender. Additional pertinent laboratory within an enclosed fascial space, which can occur after studies revealed creatine phosphokinase level of 53,350 U/L; fracture, soft tissue injury, or reperfusion after arterial isch- serum creatinine concentration had increased to 3.5 mg/dL, and emia.1 Other less common causes of compartment syn- WBC count had increased to 18,000 cells/mm3. Venous duplex drome include prolonged limb compression, burns, and scans showed no evidence of deep venous thrombosis in the right extreme exertion.1 Soft tissue infection in the form of lower leg. Pressure was measured in all four compartments of the myositis is a rare cause of compartment syndrome. We right calf and ranged from 55 to 65 mm Hg. -

Evaluation of Suspected Malignant Hyperthermia Events During Anesthesia Frank Schuster*, Stephan Johannsen, Daniel Schneiderbanger and Norbert Roewer
Schuster et al. BMC Anesthesiology 2013, 13:24 http://www.biomedcentral.com/1471-2253/13/24 RESEARCH ARTICLE Open Access Evaluation of suspected malignant hyperthermia events during anesthesia Frank Schuster*, Stephan Johannsen, Daniel Schneiderbanger and Norbert Roewer Abstract Background: Malignant hyperthermia (MH), a metabolic myopathy triggered by volatile anesthetics and depolarizing muscle relaxants, is a potentially lethal complication of general anesthesia in susceptible patients. The implementation of modern inhalation anesthetics that research indicates as less potent trigger substances and the recommended limitations of succinylcholine use, suggests there may be considerable decline of fulminant MH cases. In the presented study, the authors analyzed suspected MH episodes during general anesthesia of patients that were referred to the Wuerzburg MH unit between 2007 and 2011, assuming that MH is still a relevant anesthetic problem in our days. Methods: With approval of the local ethics committee data of patients that underwent muscle biopsy and in vitro contracture test (IVCT) between 2007 and 2011 were analyzed. Only patients with a history of suspected MH crisis were included in the study. The incidents were evaluated retrospectively using anesthetic documentation and medical records. Results: Between 2007 and 2011 a total of 124 patients were tested. 19 of them were referred because of suspected MH events; 7 patients were diagnosed MH-susceptible, 4 MH-equivocal and 8 MH-non-susceptible by IVCT. In a majority of cases masseter spasm after succinylcholine had been the primary symptom. Cardiac arrhythmias and hypercapnia frequently occurred early in the course of events. Interestingly, dantrolene treatment was initiated in a few cases only. -

The Rigid Spine Syndrome-A Myopathy of Uncertain Nosological Position
J Neurol Neurosurg Psychiatry: first published as 10.1136/jnnp.48.9.887 on 1 September 1985. Downloaded from Journal ofNeurology, Neurosurgery, and Psychiatry 1985;48:887-893 The rigid spine syndrome-a myopathy of uncertain nosological position W POEWE,* H WILLEIT,* E SLUGA,t U MAYR* From the University Clinic for Neurology, Innsbruck, * and the Neurological Instiute ofthe University of Vienna, Vienna,t Austria SUMMARY Four patients meeting the clinical criteria of the rigid spine syndrome are presented; they are one girl with a positive family history and three boys. Clinical and histological findings are discussed in relation to the 14 cases of rigid spine syndrome reported in the literature. The delineations of the syndrome from other benign myopathies with early contractures are discussed suggesting that the rigid spine syndrome probably does not represent a single nosological entity. In 1965 Dubowitz' drew attention to a muscular Case reports disorder resembling muscular dystrophy at the time guest. Protected by copyright. of its onset in infancy but of benign and non progres- Since 1978 the authors have had the opportunity to sive nature with the development of only mild examine clinically, electrophysiologically and by muscle weakness. The central clinical feature in this condi- biopsy four cases of a muscle disorder fulfilling the clinical criteria of rigid spine syndrome as described by tion is marked limitation of flexion of the cervical Dubowitz.' The patients were three males and one and dorsolumbar spine with the development of female, whose sister is thought to suffer from the same scoliosis and associated contractures of other joints, disorder. -

Electromyography and Nerve Conduction Studies in Patients With
ogy & N ol eu ur e ro N p h f y o s l Ziegler MS, J Neurol Neurophysiol 2014, 5:3 i o a l n o r g u y o DOI: 10.4172/2155-9562.1000203 J Journal of Neurology & Neurophysiology ISSN: 2155-9562 Research Article Open Access Electromyography and Nerve Conduction Studies in Patients with Lumbar Spinal Stenosis: Is Neurophysiological Examination an Important Tool? Marcus Sofia Ziegler1, Renata Siciliani Scalco2, Erasmo de Abreu Zardo1, Jefferson Becker2 and Irenio Gomes2* 1Department of Orthopaedics & Spine Surgery, Hospital São Lucas Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, Brazil 2Department of Neurology, Hospital São Lucas Pontifícia Universidade Católica do Rio Grande do Sul, Porto Alegre, Brazil *Corresponding author: Irenio Gomes, Hospital Sao Lucas, and Avenida Ipiranga 6690 - 3 ºandar - IGG Porto Alegre - RS – Brasil, CEP 90610-000, Tel: +55 (51) 3336.8153 / 3320.3000; E-mail: [email protected] Received Date: Jan 21, 2014 Accepted Date: Apr 25, 2014 Published Date: Apr 30, 2014 Copyright: © 2014 Ziegler MS, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Background: There is no single test that defines properly lumbar spinal stenosis (LSS) diagnosis, and diagnosis of the syndrome continues to rely on clinical judgment. LSS symptoms may be broad and may be seen in multiple disorders in elderly. Hypothesis: To identify the role of electromyography and nerve-conduction studies on LSS diagnosis. -
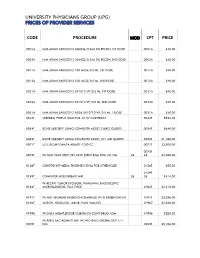
Code Procedure Cpt Price University Physicians Group
UNIVERSITY PHYSICIANS GROUP (UPG) PRICES OF PROVIDER SERVICES CODE PROCEDURE MOD CPT PRICE 0001A IMM ADMN SARSCOV2 30MCG/0.3ML DIL RECON 1ST DOSE 0001A $40.00 0002A IMM ADMN SARSCOV2 30MCG/0.3ML DIL RECON 2ND DOSE 0002A $40.00 0011A IMM ADMN SARSCOV2 100 MCG/0.5 ML 1ST DOSE 0011A $40.00 0012A IMM ADMN SARSCOV2 100 MCG/0.5 ML 2ND DOSE 0012A $40.00 0021A IMM ADMN SARSCOV2 5X1010 VP/0.5 ML 1ST DOSE 0021A $40.00 0022A IMM ADMN SARSCOV2 5X1010 VP/0.5 ML 2ND DOSE 0022A $40.00 0031A IMM ADMN SARSCOV2 AD26 5X10^10 VP/0.5 ML 1 DOSE 0031A $40.00 0042T CEREBRAL PERFUS ANALYSIS, CT W/CONTRAST 0042T $954.00 0054T BONE SURGERY USING COMPUTER ASSIST, FLURO GUIDED 0054T $640.00 0055T BONE SURGERY USING COMPUTER ASSIST, CT/ MRI GUIDED 0055T $1,188.00 0071T U/S LEIOMYOMATA ABLATE <200 CC 0071T $2,500.00 0075T 0075T PR TCAT PLMT XTRC VRT CRTD STENT RS&I PRQ 1ST VSL 26 26 $2,208.00 0126T CAROTID INT-MEDIA THICKNESS EVAL FOR ATHERSCLER 0126T $55.00 0159T 0159T COMPUTER AIDED BREAST MRI 26 26 $314.00 PR RECTAL TUMOR EXCISION, TRANSANAL ENDOSCOPIC 0184T MICROSURGICAL, FULL THICK 0184T $2,315.00 0191T PR ANT SEGMENT INSERTION DRAINAGE W/O RESERVOIR INT 0191T $2,396.00 01967 ANESTH, NEURAXIAL LABOR, PLAN VAG DEL 01967 $2,500.00 01996 PR DAILY MGMT,EPIDUR/SUBARACH CONT DRUG ADM 01996 $285.00 PR PERQ SAC AGMNTJ UNI W/WO BALO/MCHNL DEV 1/> 0200T NDL 0200T $5,106.00 PR PERQ SAC AGMNTJ BI W/WO BALO/MCHNL DEV 2/> 0201T NDLS 0201T $9,446.00 PR INJECT PLATELET RICH PLASMA W/IMG 0232T HARVEST/PREPARATOIN 0232T $1,509.00 0234T PR TRANSLUMINAL PERIPHERAL ATHERECTOMY, RENAL -

Compartment Syndrome
Rowan University Rowan Digital Works Stratford Campus Research Day 23rd Annual Research Day May 2nd, 12:00 AM A Case of Atraumatic Posterior Thigh Compartment Syndrome Nailah Mubin Rowan University Brian Katt M.D. Rothman Institute Follow this and additional works at: https://rdw.rowan.edu/stratford_research_day Part of the Cardiovascular Diseases Commons, Hemic and Lymphatic Diseases Commons, Nephrology Commons, Orthopedics Commons, Pathological Conditions, Signs and Symptoms Commons, and the Surgery Commons Let us know how access to this document benefits ouy - share your thoughts on our feedback form. Mubin, Nailah and Katt, Brian M.D., "A Case of Atraumatic Posterior Thigh Compartment Syndrome" (2019). Stratford Campus Research Day. 47. https://rdw.rowan.edu/stratford_research_day/2019/may2/47 This Poster is brought to you for free and open access by the Conferences, Events, and Symposia at Rowan Digital Works. It has been accepted for inclusion in Stratford Campus Research Day by an authorized administrator of Rowan Digital Works. A Case of Atraumatic Posterior Thigh Compartment Syndrome Nailah Mubin OMS-III1, Brian Katt MD2 1Rowan University School of Osteopathic Medicine, 2Brielle Orthopedics at Rothman Institute Introduction Hospital Course Compartment syndrome (CS): intra-compartmental pressures exceed Day 1 4:40am Day 1 5pm Day 4 Day 5 Day 9 Day 12 Day 16 Day 18 Day 19 to a point where arterial, venous and lymphatic circulation of local 1 tissues, muscles and nerves is compromised ER Admit Fasciotomy Cr=7.54 First Closure Pt reports Complete Cr=5.25 Cr=4.09 Discharge 2 • Most common after a traumatic injury . Usually occurs in the leg Dialysis Initiated Attempt increased sensation Closure Cleared by Nephro or forearm and less commonly in the thigh3 • Thigh compartment syndrome (TCS) is rare due to its larger size Surgical Intervention Discussion and more compliant borders. -

Why Electromyoneurography Instead Of
Neurological Disorders and Therapeutics Research Article ISSN: 2514-4790 Why electromyoneurography instead of electromyography and electroneurography? Electroneurography is essential for interpretation of electromyographic results! Anica Jušić* Shool of Medicine University of Zagreb, Zagreb, 10000 Zagreb, Gundulićeva 49, Croatia Abstract The author suggests revival and further development of old method who had proven to be of significant benefit in differential dignostics of nerve lesions and displayed significant research possibilities. The basic idea is the unification of electromyographic results with neural stimulation. Therefore the author suggests again, to use new name - Electromyoneurography for the old methods. Introduction electrodes as I learned at Albrecht Struppler’s laboratory. The stimulation may be done with surface electrodes also, but the reliability, George Bernard Shaw once wrote: „The single biggest problem constancy and reproducibility of the results, with such technique, in communication is the illusion that it has taken place“. I hope this decreases significantly. With needle electrodes, by simple switching introductory part will make possible, among nowadays achieved of the polarity, besides the efferent motor conduction velocity the scientific circumstances, usage and further development of methods additional afferent conduction velocity measurements can be done. If differentiated some decades ago in Centre /Institute for neuromuscular necessary, the ribbon electrodes for percutaneous evocation of sensory disease, of University Hospital Clinic Zagreb, which I have founded, potentials may be involved. 1973. The purpose of this review is to shed the light on the old methods who had proven to be of significant benefit in differential dignosis of Analyses of evoked muscle or nerve potentials clarifyes so many nerve lesions and possible basis for further scientific research. -

Icd-9-Cm (2010)
ICD-9-CM (2010) PROCEDURE CODE LONG DESCRIPTION SHORT DESCRIPTION 0001 Therapeutic ultrasound of vessels of head and neck Ther ult head & neck ves 0002 Therapeutic ultrasound of heart Ther ultrasound of heart 0003 Therapeutic ultrasound of peripheral vascular vessels Ther ult peripheral ves 0009 Other therapeutic ultrasound Other therapeutic ultsnd 0010 Implantation of chemotherapeutic agent Implant chemothera agent 0011 Infusion of drotrecogin alfa (activated) Infus drotrecogin alfa 0012 Administration of inhaled nitric oxide Adm inhal nitric oxide 0013 Injection or infusion of nesiritide Inject/infus nesiritide 0014 Injection or infusion of oxazolidinone class of antibiotics Injection oxazolidinone 0015 High-dose infusion interleukin-2 [IL-2] High-dose infusion IL-2 0016 Pressurized treatment of venous bypass graft [conduit] with pharmaceutical substance Pressurized treat graft 0017 Infusion of vasopressor agent Infusion of vasopressor 0018 Infusion of immunosuppressive antibody therapy Infus immunosup antibody 0019 Disruption of blood brain barrier via infusion [BBBD] BBBD via infusion 0021 Intravascular imaging of extracranial cerebral vessels IVUS extracran cereb ves 0022 Intravascular imaging of intrathoracic vessels IVUS intrathoracic ves 0023 Intravascular imaging of peripheral vessels IVUS peripheral vessels 0024 Intravascular imaging of coronary vessels IVUS coronary vessels 0025 Intravascular imaging of renal vessels IVUS renal vessels 0028 Intravascular imaging, other specified vessel(s) Intravascul imaging NEC 0029 Intravascular -

The Hypotonic Infant: Clinical Approach
Journal of Pediatric Neurology 5 (2007) 181–187 181 IOS Press Review Article The hypotonic infant: Clinical approach Mohammed M.S. Jan∗ Department of Pediatrics, King Abdulaziz University Hospital, and Department of Neurosciences, King Faisal Specialist Hospital & RC, Jeddah, Saudi Arabia Received 27 November 2006 Revised 25 December 2006 Accepted 31 December 2006 Abstract. Hypotonia in infants can be a confusing clinical presentation leading to inaccurate evaluation and unnecessary investigations. Hypotonia can result from a variety of central or peripheral causes. Therefore, hypotonia is a phenotype of many clinical conditions with variable prognosis. It is important to recognize that hypotonia is not equivalent to weakness. Infants with central causes, such as Down syndrome, may have severe hypotonia with normal muscle strength. Peripheral hypotonia is frequently associated with weakness, which can be predominantly distal in neuropathies or predominantly proximal in myopathies. In general, central hypotonia is much more commonly encountered; however, the prognosis is worst for hypotonia secondary to neuromuscular pathology. The distinction between central and peripheral hypotonia is therefore critical for proper evaluation and management. Stepwise and accurate assessment is very important to reach the correct diagnosis promptly. In this review, I present a concise clinical approach for evaluating the hypotonic infant. Some practical tips and skills are discussed to improve the likelihood of obtaining an accurate diagnosis. Reaching a specific diagnosis is needed for providing appropriate therapy, prognosis, and counseling. Keywords: Infant, child, hypotonia, floppy, examination, approach 1. Introduction rological disorders one of the most difficult aspects of their clinical practice [6–8]. Hypotonia in infants Neurological disorders are common in Saudi Ara- and children can be a confusing clinical presentation, bia accounting for 25–30% of all consultations to pe- which often leads to inaccurate evaluation and unnec- diatrics [1]. -

Bryce Macek Supervisors
Exploring the Biologics of Rotator CuffInjury and Advancing Repair Student: Bryce Macek Supervisors: Dr. Jeff Leiter, Dr. Peter MacDonald Department of Orthopedic Surgery Rotator cufftears are a common problem associated with muscle atrophy and fatty infiltration. These changes may be progressive and even irreversible despite successful repair. A deeper understanding of the cellular processes contributing to these degenerative changes is needed to predict outcomes. The objectives of the present study are to: 1) characterize rotator cuff tears through clinical exam and MRI, 2) compare muscle atrophy at the cellular level via muscle biopsy of torn supraspinatus and deltoid, 3) determine if cuff tear size is related to clinical variables. Ten patients with clinical and MRI evidence of a rotator cuff tear were biopsied from supraspinatus and deltoid muscles during arthroscopy. Samples were stained with hematoxylin and eosin to determine fiber diameter. Fiber diameter of the deltoid muscle was greater than the supraspinatus (p<.OOl). Distribution of fiber diameter of the supraspinatus and deltoid muscles did not follow a normal distribution, which may indicate muscle atrophy. The deltoid muscle of seven patients did follow a normal distribution compared to three supraspinatus muscles. The results of this study suggest that the deltoid muscle is a viable option to use as a control in microscopic studies of rotator cuff muscles. When combined with the other phases of this research project the results ofthis study have the potential to provide insight into the mechanisms responsible for muscle atrophy and fatty infiltration in shoulder injury. This information can be used to guide new treatments and increase the effectiveness of current interventions. -

ICD-9-CM Coordination and Maintenance Committee Meeting September 28-29, 2006 Diagnosis Agenda
ICD-9-CM Coordination and Maintenance Committee Meeting September 28-29, 2006 Diagnosis Agenda Welcome and announcements Donna Pickett, MPH, RHIA Co-Chair, ICD-9-CM Coordination and Maintenance Committee ICD-9-CM TIMELINE .................................................................................................... 2 Hearing loss, speech, language, and swallowing disorders ........................................... 8 Kyle C. Dennis, Ph.D., CCC-A, FAAA and Dee Adams Nikjeh,Ph.D., CCC-SLP American Speech-Language-Hearing Association Urinary risks factors for bladder cancer ...................................................................... 13 Louis S. Liou, M.D., Ph.D., Abbott Chronic Total Occlusion of Artery of Extremities....................................................... 15 Matt Selmon, M.D., Cordis Osteonecrosis of jaw ....................................................................................................... 17 Vincent DiFabio,M.D., American Association of Oral and Maxillofacial Surgeons Intraoperative Floppy Iris Syndrome ........................................................................... 18 Priscilla Arnold, M.D., American Society of Cataract and Refractive Surgery Septic embolism............................................................................................................... 19 Parvovirus B19 ................................................................................................................ 21 Avian Influenza (Bird Flu)............................................................................................ -

Evaluating the Patient with Suspected Radiculopathy
EVALUATINGEVALUATING THETHE PATIENTPATIENT WITHWITH SUSPECTEDSUSPECTED RADICULOPATHYRADICULOPATHY Timothy R. Dillingham, M.D., M.S Professor and Chair, Department of Physical Medicine and Rehabilitation The Medical College of Wisconsin. RadiculopathiesRadiculopathies PathophysiologicalPathophysiological processesprocesses affectingaffecting thethe nervenerve rootsroots VeryVery commoncommon reasonreason forfor EDXEDX referralreferral CAUSESCAUSES OFOF RADICULOPATHYRADICULOPATHY HNPHNP RadiculiitisRadiculiitis SpinalSpinal StenosisStenosis SpondylolisthesisSpondylolisthesis InfectionInfection TumorTumor FacetFacet SynovialSynovial CystCyst Diseases:Diseases: Diabetes,Diabetes, AIDPAIDP MUSCULOSKELETALMUSCULOSKELETAL DISORDERSDISORDERS :: UPPERUPPER LIMBLIMB ShoulderShoulder BursitisBursitis LateralLateral EpicondylitisEpicondylitis DequervainsDequervains TriggerTrigger fingerfinger FibrositisFibrositis FibromyalgiaFibromyalgia // regionalregional painpain syndromesyndrome NEUROLOGICALNEUROLOGICAL CONDITIONSCONDITIONS MIMICKINGMIMICKING CERVICALCERVICAL RADICULOPATHYRADICULOPATHY Entrapment/CompressionEntrapment/Compression neuropathiesneuropathies –– Median,Median, Radial,Radial, andand UlnarUlnar BrachialBrachial NeuritisNeuritis MultifocalMultifocal MotorMotor NeuropathyNeuropathy NeedNeed ExtensiveExtensive EDXEDX studystudy toto R/OR/O otherother conditionsconditions MUSCULOSKELETALMUSCULOSKELETAL DISORDERSDISORDERS :: LOWERLOWER LIMBLIMB HipHip arthritisarthritis TrochantericTrochanteric BursitisBursitis IlliotibialIlliotibial BandBand