The Genome of Avibacterium Paragallinarum Bacterium
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bacterial Communities of the Upper Respiratory Tract of Turkeys
www.nature.com/scientificreports OPEN Bacterial communities of the upper respiratory tract of turkeys Olimpia Kursa1*, Grzegorz Tomczyk1, Anna Sawicka‑Durkalec1, Aleksandra Giza2 & Magdalena Słomiany‑Szwarc2 The respiratory tracts of turkeys play important roles in the overall health and performance of the birds. Understanding the bacterial communities present in the respiratory tracts of turkeys can be helpful to better understand the interactions between commensal or symbiotic microorganisms and other pathogenic bacteria or viral infections. The aim of this study was the characterization of the bacterial communities of upper respiratory tracks in commercial turkeys using NGS sequencing by the amplifcation of 16S rRNA gene with primers designed for hypervariable regions V3 and V4 (MiSeq, Illumina). From 10 phyla identifed in upper respiratory tract in turkeys, the most dominated phyla were Firmicutes and Proteobacteria. Diferences in composition of bacterial diversity were found at the family and genus level. At the genus level, the turkey sequences present in respiratory tract represent 144 established bacteria. Several respiratory pathogens that contribute to the development of infections in the respiratory system of birds were identifed, including the presence of Ornithobacterium and Mycoplasma OTUs. These results obtained in this study supply information about bacterial composition and diversity of the turkey upper respiratory tract. Knowledge about bacteria present in the respiratory tract and the roles they can play in infections can be useful in controlling, diagnosing and treating commercial turkey focks. Next-generation sequencing has resulted in a marked increase in culture-independent studies characterizing the microbiome of humans and animals1–6. Much of these works have been focused on the gut microbiome of humans and other production animals 7–11. -

Identification of Pasteurella Species and Morphologically Similar Organisms
UK Standards for Microbiology Investigations Identification of Pasteurella species and Morphologically Similar Organisms Issued by the Standards Unit, Microbiology Services, PHE Bacteriology – Identification | ID 13 | Issue no: 3 | Issue date: 04.02.15 | Page: 1 of 28 © Crown copyright 2015 Identification of Pasteurella species and Morphologically Similar Organisms Acknowledgments UK Standards for Microbiology Investigations (SMIs) are developed under the auspices of Public Health England (PHE) working in partnership with the National Health Service (NHS), Public Health Wales and with the professional organisations whose logos are displayed below and listed on the website https://www.gov.uk/uk- standards-for-microbiology-investigations-smi-quality-and-consistency-in-clinical- laboratories. SMIs are developed, reviewed and revised by various working groups which are overseen by a steering committee (see https://www.gov.uk/government/groups/standards-for-microbiology-investigations- steering-committee). The contributions of many individuals in clinical, specialist and reference laboratories who have provided information and comments during the development of this document are acknowledged. We are grateful to the Medical Editors for editing the medical content. For further information please contact us at: Standards Unit Microbiology Services Public Health England 61 Colindale Avenue London NW9 5EQ E-mail: [email protected] Website: https://www.gov.uk/uk-standards-for-microbiology-investigations-smi-quality- and-consistency-in-clinical-laboratories UK Standards for Microbiology Investigations are produced in association with: Logos correct at time of publishing. Bacteriology – Identification | ID 13 | Issue no: 3 | Issue date: 04.02.15 | Page: 2 of 28 UK Standards for Microbiology Investigations | Issued by the Standards Unit, Public Health England Identification of Pasteurella species and Morphologically Similar Organisms Contents ACKNOWLEDGMENTS ......................................................................................................... -

Pasteurellaceae: P. Multocida, Avibacterium Gallinarum, A
Pasteurellaceae: P. Multocida, Avibacterium gallinarum, A. paragallinarum All the members of the family Pasteurellaceae are gram negative coccobacilli. They are facultative anaerobes, and typically oxidase-positive (which sets them apart from members of the family Enterobacteriaceae). Morphology and Staining Members of the genera Avibacterium, and Pasteurella are gram-negative coccobacilli. Bipolarity, that is, the staining of only the tips of cells, may be demonstrable with polychrome stains (e.g., Wright’s stain). Cell structure Adhesins. Some and probably all members of the family Pasteurellaceae produce adhesins (and possibly more than one kind). A type 4 fimbria (adhesin) has been described for avian strains of P. Multocida. Capsule. The hyaluronic acid capsule of type A strains P. multocida serves as an adhesin. The hyaluronic acid is similar (if not identical) to host tissue components, and is thus poorly antigenic; they also bind complement components poorly (and is therefore antiphagocytic).The hyaluronic acid capsule also serves as an adhesin for respiratory tract epithelial cells as in the case of capsule type A strains of P. Multocida Exotoxin. Pasteurella produce a number of proteins with toxic activity. At least two of these are important in the pathogenesis of disease: RTX and Rho toxin Growth Characteristics Avibacterium and Pasteurella grow best in the presence of serum or blood. After overnight incubation (35–37 ◦C), colonies are up to 2 mm in diameter, clear to 1 grayish, and smooth or mucoid. All are gram-negative, nonmotile coccobacilli. They are facultative anaerobes, typically oxidase-positive. Variability P. multocida consists of 5 capsular serogroups (A, B, D, E, and F) and 16 somatic serotypes (1–16). -
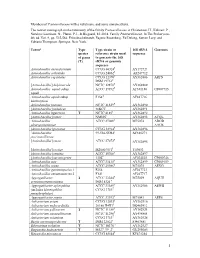
Members of Pasteurellaceae with a Valid Name and Some Unnamed Taxa
Members of Pasteurellaceae with a valid name and some unnamed taxa. The newest monograph on the taxonomy of the family Pasteurellaceae is Christensen, H., Kuhnert, P., Nørskov-Lauritsen, N., Planet, P.J., & Bisgaard, M. 2014. Family Pasteurellaceae. In The Prokaryotes 4th ed. Vol. 9, pp. 535-564. Erko Stackebrandt, Eugene Rosenberg, Ed Delong, Steven Lory, and Fabiano Thompson. Springer, New York. Taxona Type Type strain or 16S rRNA Genomes species reference strain used sequence of genus to generate the 16S (T) rRNA or genomic sequence Actinobacillus anseriformium CCUG 60324T AY172727 Actinobacillus arthritidis CCUG 24862T AF247712 Actinobacillus capsulatus CCUG 12396T AY362886 ARFN DSM 19761T [Actinobacillus] delphinicola NCTC 12870T AY362889 Actinobacillus equuli subsp. ATCC 19392T AF381186 CP007715 equuli Actinobacillus equuli subsp. F154T AF247716 haemolyticus Actinobacillus hominis NCTC 11529T AY362890 [Actinobacillus] indolicus 46KC2T AY362891 Actinobacillus lignieresii T NCTC 4189T AY362892 [Actinobacillus] minor NM305T AY362893 ACQL Actinobacillus ATCC 27088T M75074 ADOD pleuropneumoniae AACK [Actinobacillus] porcinus CCUG 38924T AY362896 ‘Actinobacillus 99-536-55H-F AF486274 porcitonsillarum’ T [Actinobacillus] rossii ATCC 27072 AY362895 [Actinobacillus] scotiae M2000/95/1T Y09653 [Actinobacillus] seminis ATCC 15768T AY362897 [Actinobacillus] succinogenes 130ZT AF024525 CP000746 Actinobacillus suis ATCC 33415T AY362899 CP009159 Actinobacillus ureae ATCC 25986T M75075 AEVG Actinobacillus genomospecies 1 F264 AF247723 Actinobacillus -

Data-Mining of Antibiotic Resistance Genes Provides Insight Into the Community
bioRxiv preprint doi: https://doi.org/10.1101/246033; this version posted January 10, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 1 Data-mining of Antibiotic Resistance Genes Provides Insight into the Community 2 Structure of Ocean Microbiome 3 Shiguang Hao1,$, Pengshuo Yang1,$, Maozhen Han1,$, Junjie Xu1, Shaojun Yu1, Chaoyun 4 Chen1, Wei-Hua Chen1, Houjin Zhang1,*, Kang Ning1,* 5 1Key Laboratory of Molecular Biophysics of the Ministry of Education, College of Life 6 Science and Technology, Huazhong University of Science and Technology, Wuhan, Hubei, 7 430074, China 8 9 $ These authors contributed equally to this work. 10 * Corresponding author. E-mail: [email protected], [email protected]. 11 1 bioRxiv preprint doi: https://doi.org/10.1101/246033; this version posted January 10, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. 12 Abstract 13 Background:Antibiotics have been spread widely in environments, asserting profound 14 effects on environmental microbes as well as antibiotic resistance genes (ARGs) within these 15 microbes. Therefore, investigating the associations between ARGs and bacterial communities 16 become an important issue for environment protection. Ocean microbiomes are potentially 17 large ARG reservoirs, but the marine ARG distribution and its associations with bacterial 18 communities remain unclear. -

Alternatives to Antibiotics: a Symposium on the Challenges and Solutions for Animal Health and Production
antibiotics Conference Report Alternatives to Antibiotics: A Symposium on the Challenges and Solutions for Animal Health and Production Todd R. Callaway 1,* , Hyun Lillehoj 2, Rungtip Chuanchuen 3 and Cyril G. Gay 4 1 Department of Animal and Dairy Science, University of Georgia, Athens, GA 30602, USA 2 Beltsville Agricultural Research Center, Animal Bioscience and Biotechnology Laboratory, USDA-Agricultural Research Service, Beltsville, MD 20705, USA; [email protected] 3 Research Unit in Microbial Food Safety and Antimicrobial Resistance, Department of Veterinary Public Health, Faculty of Veterinary Science, Chulalongkorn University, Bangkok 10330, Thailand; [email protected] 4 Office of National Programs, USDA-Agricultural Research Service, Beltsville, MD 20705, USA; [email protected] * Correspondence: [email protected]; Tel.: +1-706-542-0962 Abstract: Antibiotics have improved the length and quality of life of people worldwide and have had an immeasurable influence on agricultural animal health and the efficiency of animal production over the last 60 years. The increased affordability of animal protein for a greater proportion of the global population, in which antibiotic use has played a crucial part, has resulted in a substantial improvement in human quality of life. However, these benefits have come with major unintended consequences, including antibiotic resistance. Despite the inherent benefits of restricting antibiotic use in animal production, antibiotics remain essential to ensuring animal health, necessitating the -
![Reclassification of Pasteurella Gallinarum, [Haemophilus]](https://docslib.b-cdn.net/cover/9354/reclassification-of-pasteurella-gallinarum-haemophilus-6899354.webp)
Reclassification of Pasteurella Gallinarum, [Haemophilus]
International Journal of Systematic and Evolutionary Microbiology (2005), 55, 353–362 DOI 10.1099/ijs.0.63357-0 Reclassification of Pasteurella gallinarum, [Haemophilus] paragallinarum, Pasteurella avium and Pasteurella volantium as Avibacterium gallinarum gen. nov., comb. nov., Avibacterium paragallinarum comb. nov., Avibacterium avium comb. nov. and Avibacterium volantium comb. nov. Patrick J. Blackall,1 Henrik Christensen,2 Tim Beckenham,3 Linda L. Blackall3 and Magne Bisgaard2 Correspondence 1Department of Primary Industries and Fisheries, Animal Research Institute, Yeerongpilly, Patrick J. Blackall Queensland 4105, Australia [email protected] 2Department of Veterinary Pathobiology, The Royal Veterinary and Agricultural University, Stigbøjlen 4, 1870 Frederiksberg C, Denmark 3Department of Microbiology and Parasitology, School of Molecular and Microbial Sciences, The University of Queensland, St Lucia, Queensland 4072, Australia This paper describes a phenotypic and genotypic investigation of the taxonomy of [Haemophilus] paragallinarum, Pasteurella gallinarum, Pasteurella avium and Pasteurella volantium, a major subcluster within the avian 16S rRNA cluster 18 of the family Pasteurellaceae. An extended phenotypic characterization was performed of the type strain of [Haemophilus] paragallinarum, which is NAD-dependent, and eight NAD-independent strains of [Haemophilus] paragallinarum. Complete 16S rRNA gene sequences were obtained for one NAD-independent and four NAD-dependent [Haemophilus] paragallinarum strains. These five sequences along with existing 16S rRNA gene sequences for 11 other taxa within avian 16S rRNA cluster 18 as well as seven other taxa from the Pasteurellaceae were subjected to phylogenetic analysis. The analysis demonstrated that [Haemophilus] paragallinarum, Pasteurella gallinarum, Pasteurella avium and Pasteurella volantium formed a monophyletic group with a minimum of 96?8 % sequence similarity. -

Avian Infectious Bronchitis Coronavirus Variation and Selection
Avian Infectious Bronchitis Coronavirus Variation and Selection by Rodrigo A. Gallardo A dissertation submitted to the Graduate Faculty of Auburn University in partial fulfillment of the requirements for the Degree of Doctor of Philosophy Auburn, Alabama December 12, 2011 Keywords: Infectious bronchitis virus, coronavirus evolution, chicken, genetic variation, pathology, transmission Copyright 2011 by Rodrigo A. Gallardo Approved by Haroldo Toro, Chair, Professor of Pathobiology Vicky L. van Santen Co-chair, Professor of Pathobiology Frederic J. Hoerr, Professor of Pathobiology Stuart B. Price, Associate Professor of Pathobiology Sharon R. Roberts, Associate Professor of Biological Sciences Abstract Infectious bronchitis virus (IBV) is a highly variable virus, its genetic variation is generated by nucleotide insertions, deletions, or point mutations made by the viral polymerase during virus replication. Another mechanism for IBV variability is RNA recombination. Selection of IBV in the host has also been previously demonstrated. Specific objectives during this three-part investigation were to assess IBV intraspatial variation in chickens, determine the IBV evolutionary pathway in viral immunodeficient hosts, and evaluate IBV variation and effects on testes of young and adult roosters. Variation inside the chicken (Intraspatial variation) was assessed, we inoculated chickens with an Ark-type IBV commercial vaccine and characterized the sequences of the spike (S) 1 gene of IBV contained in tear fluid, trachea, and reproductive tract. The predominant IBV phenotype contained in the vaccine (prior to inoculation), became a minor or non-detectable population at all times in all tissues after replication in the chickens. Five new predominant populations designated as component (C) 1 through C5, showing distinct non-synonymous changes, were detected in the tissues or fluids of individual vaccinated chickens. -

340109-7.Pdf
Table S3. DESeq results: ESVs from the skin that were identified as significantly enriched in either non-ectoparasitized (positive log2FoldChange values; black or ectoparasitized (negative log2FoldChange values; red) bats. Host_family baseMean log2FoldChange lfcSE stat pvalue padj Rank1 Rank2 Rank3 Rank4 Rank5 Rank6 Rank7 Hipposideridae 33.3360233 -25.65717963 2.84165626 -9.0289526 1.73E-19 4.77E-17 Bacteria Bacteroidetes [Saprospirae] [Saprospirales] Saprospiraceae Unclassified Unclassified Hipposideridae 24.9053051 -25.44743324 2.961084869 -8.5939561 8.40E-18 1.54E-15 Bacteria Proteobacteria Gammaproteobacteria Salinisphaerales Salinisphaeraceae Salinisphaera Unclassified Hipposideridae 21.6441823 -25.2429634 2.961117027 -8.5248111 1.53E-17 2.68E-15 Bacteria Bacteroidetes [Rhodothermi] [Rhodothermales] [Balneolaceae] KSA1 Unclassified Hipposideridae 35.4879494 -25.2388568 2.420732479 -10.426124 1.88E-25 3.63E-22 Bacteria Firmicutes Clostridia Clostridiales Clostridiaceae Unclassified Unclassified Hipposideridae 20.5283726 -25.17048193 2.799904489 -8.9897645 2.48E-19 6.36E-17 Archaea Euryarchaeota Halobacteria Halobacteriales Halobacteriaceae Natronomonas Unclassified Hipposideridae 33.6261871 -24.50404309 2.849792516 -8.5985358 8.07E-18 1.54E-15 Bacteria Bacteroidetes Flavobacteriia Flavobacteriales Flavobacteriaceae Unclassified Unclassified Hipposideridae 11.6313092 -24.40177521 2.306373551 -10.580149 3.68E-26 1.42E-22 Bacteria Proteobacteria Gammaproteobacteria Salinisphaerales Salinisphaeraceae Salinisphaera Unclassified Hipposideridae -

ISOLATION and IDENTIFICATION of Avibacterium Paragallinarum from LAYER CHICKENS
ISOLATION AND IDENTIFICATION OF Avibacterium paragallinarum FROM LAYER CHICKENS A THESIS Submitted to Bangladesh Agricultural University, Mymensingh In partial Fulfillment of the Requirements for the Degree of Master of Science in Pathology BY SALEHA AKTER Roll No.: 11Vet Path JJ 14 M Registration No.: 38019, Session: 20011-12 Department of Pathology Faculty of Veterinary Science Bangladesh Agricultural University Mymensingh May 2012 ISOLATION AND IDENTIFICATION OF Avibacterium paragallinarum FROM LAYER CHICKENS A THESIS Submitted to Bangladesh Agricultural University, Mymensingh In partial Fulfillment of the Requirements for the Degree of Master of Science in Pathology By SALEHA AKTER Approved as to style and content by Prof. Dr. Priya Mohan Das Prof. Dr. Md. Mokbul Hossain Co- Supervisor Supervisor Prof. Dr. Priya Mohan Das Chairman, BOS & Head Department of Pathology May, 2012 ACKNOWLEDGEMENTS The author is grateful and indebted to the almighty Allah without whose grace she would have ever been able to pursue her higher studies in this field of science and to complete her thesis for the degree of Master of Science (MS) in Pathology. The author expresses her ever indebtedness, deepest sense of gratitude, sincere appreciation and profound regards to her reverend and beloved teacher and research Supervisor, Professor Dr. Md.Mokbul Hossian, Department of Pathology Bangladesh Bangladesh Agricultural University, (BAU) Mymensingh for his scholastic guidance, uncompromising principles, sympathetic supervision, valuable advice, constant inspiration, affectionate feeling, radical investigation and constructive criticism in all phases of this study and preparing the manuscript. The author finds it a great pleasure in expressing her heartfelt gratitude and immense indebtedness to her honourable and respected Research Co –Supervisor Dr. -

Pasteurellaceae Members with Similar Morphological Patterns Associated with Respiratory Manifestations in Ducks
Veterinary World, EISSN: 2231-0916 RESEARCH ARTICLE Available at www.veterinaryworld.org/Vol.12/December-2019/24.pdf Open Access Pasteurellaceae members with similar morphological patterns associated with respiratory manifestations in ducks Samah Eid1, Sherif Marouf2, Hefny Y. Hefny3 and Nayera M. Al-Atfeehy1 1. Department of Bacteriology, Reference Laboratory for Veterinary Quality Control on Poultry Production, Animal Health Research Institute, Agricultural Research Centre, Nadi El-Seid St., P.O. Box 246, Dokki, Giza 12618, Egypt; 2. Department of Microbiology, Faculty of Veterinary Medicine, Cairo University, Giza, Egypt; 3. Department of Poultry Diseases, Zagazig Provincial Laboratory, Animal Health Research Institute, Agricultural Research Centre, Sharkia, Egypt. Corresponding author: Samah Eid, e-mail: [email protected] Co-authors: SM: [email protected], HYH: [email protected], NMA: [email protected] Received: 30-08-2019, Accepted: 25-11-2019, Published online: 26-12-2019 doi: www.doi.org/10.14202/vetworld.2019.2061-2069 How to cite this article: Eid S, Marouf S, Hefny HY, Al-Atfeehy NM (2019) Pasteurellaceae members with similar morphological patterns associated with respiratory manifestations in ducks, Veterinary World, 12(12): 2061-2069. Abstract Aim: A total of 112 freshly dead ducks aged from 2 to 20 weeks old with a history of respiratory manifestations were investigated for the implication of Pasteurellaceae family members. Materials and Methods: Isolation and identification to the family level were conducted by conventional bacteriological methods, including microscopic examination and biochemical characterization. Identification to the species level was conducted by polymerase chain reaction (PCR) and analytical profile index (API) 20E kits. Results: Conventional bacteriological isolation and biochemical characterization revealed the infection of 16/112 examined birds with a prevalence rate of 14.3%. -

Dynamics of Bacterial and Fungal Communities Associated with Eggshells During Incubation Stephanie� Grizard1,2, Francisco Dini-Andreote2, B
Dynamics of bacterial and fungal communities associated with eggshells during incubation Stephanie Grizard1,2, Francisco Dini-Andreote2, B. Irene Tieleman1 & Joana F. Salles2 1Department of Animal Ecology, Centre for Ecological and Evolutionary Studies, University of Groningen, Nijenborgh 7, Groningen NL-9747 AG, The Netherlands 2Department of Microbial Ecology, Centre for Ecological and Evolutionary Studies, University of Groningen, Nijenborgh 7, Groningen NL-9747 AG, The Netherlands Keywords Abstract Birds, eggshells, incubation, microbes, molecular tools. Microorganisms are closely associated with eggs and may play a determinant role in embryo survival. Yet, the majority of studies focusing on this association Correspondence relied on culture-based methodology, eventually leading to a skewed assessment Stephanie Grizard, PO Box 11103, Groningen of microbial communities. By targeting the 16S rRNA gene and internal tran- 9700 CC, The Netherlands. Tel: +31(0)50- scribed spacer (ITS) region, we, respectively, described bacterial and fungal 363-2169; E-mail: [email protected] communities on eggshells of the homing pigeon Columba livia. We explored their structure, abundance, and composition. Firstly, we showed that sampling Funding Information This work was supported by a Vidi grant technique affected the outcome of the results. While broadly used, the egg from the Netherlands Organisation for swabbing procedure led to a lower DNA extraction efficiency and provided dif- Scientific Research (to BIT). ferent profiles of bacterial communities than those based on crushed eggshell pieces. Secondly, we observed shifts in bacterial and fungal communities during Received: 28 January 2014; Accepted: 30 incubation. At late incubation, bacterial communities showed a reduction in January 2014 diversity, while their abundance increased, possibly due to the competitive advantage of some species.