Effects of Antibiotics on the Bacterial Community, Metabolic Functions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Discovered by Genomics Putative Reductive Dehalogenases with N-Terminus Transmembrane Helixes
Discovered by genomics putative reductive dehalogenases with N-terminus transmembrane helixes Atashgahi, S. This is a "Post-Print" accepted manuscript, which has been published in "FEMS microbiology ecology" This version is distributed under a non-commercial no derivatives Creative Commons (CC-BY-NC-ND) user license, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited and not used for commercial purposes. Further, the restriction applies that if you remix, transform, or build upon the material, you may not distribute the modified material. Please cite this publication as follows: Atashgahi, S. (2019). Discovered by genomics putative reductive dehalogenases with N-terminus transmembrane helixes. FEMS microbiology ecology, 95(5), [fiz048]. https://doi.org/10.1093/femsec/fiz048 1 Discovered by genomics: Putative reductive dehalogenases with N-terminus 2 transmembrane helixes 3 Siavash Atashgahi 1,2,3 4 1 Laboratory of Microbiology, Wageningen University & Research, Wageningen, The 5 Netherlands 6 2 Department of Microbiology, Radboud University, Nijmegen, The Netherlands 7 3 Soehngen Institute of Anaerobic Microbiology, Nijmegen, The Netherlands 8 [email protected]; [email protected]; http://orcid.org/0000-0002-2793- 9 2321; +31243652564 10 11 Abstract 12 Attempts for bioremediation of toxic organohalogens resulted in the identification 13 of organohalide-respiring bacteria harbouring reductive dehalogenases (RDases) enzymes. 14 RDases consist of the catalytic subunit (RdhA, encoded by rdhA) that does not have 15 membrane-integral domains, and a small putative membrane anchor (RdhB, encoded by 16 rdhB) that (presumably) locates the A subunit to the outside of the cytoplasmic membrane. 17 Recent genomic studies identified a putative rdh gene in an uncultured deltaproteobacterial 18 genome that was not accompanied by an rdhB gene, but contained transmembrane helixes 19 in N-terminus. -

Carboxylicivirga Flava Sp. Nov., Isolated from Marine Surface Sediment Hui Wang,1 Cancan Qi,1 Weiwei Chen,1 Wenwen Dong,1 Haitian Tang2 and Xiaoke Hu1
International Journal of Systematic and Evolutionary Microbiology (2016), 66, 5412–5416 DOI 10.1099/ijsem.0.001533 Carboxylicivirga flava sp. nov., isolated from marine surface sediment Hui Wang,1 Cancan Qi,1 Weiwei Chen,1 Wenwen Dong,1 Haitian Tang2 and Xiaoke Hu1 Correspondence 1Key Laboratory of Coastal Biology and Bioresource Utilization, Yantai Institute of Costal Zone Xiaoke Hu Research, Chinese Academy of Sciences, Yantai 264003, PR China [email protected] 2Yantai Marine Environment Monitoring Central Station, State Oceanic Administration, Yantai 264006, PR China A novel bacterial strain, designated Q15T, was isolated from sediments obtained from the Bohai Sea in China and subjected to a polyphasic taxonomic study. Cells of strain Q15T were Gram- stain-negative, strictly aerobic rods that produced circular, flat, orange colonies. Phylogenetic analysis based on 16S rRNA gene sequences revealed that Q15T was affiliated with the genus Carboxylicivirga in the family Marinilabiliaceae of the phylum Bacteroidetes. Strain Q15T differed genotypically from the type strains of the three recognized species of this genus (Carboxylicivirga taeanensis MEBiC 08903T, Carboxylicivirga mesophila MEBiC 07026T and Carboxylicivirga linearis FB218T) and shared 94.0–95.2 % 16S rRNA gene sequence similarity with them. The DNA G+C content of strain Q15T was 44.7 mol%. The predominant cellular fatty acids were iso-C15 : 0, anteiso-C15 : 0 and iso-C17 : 0 3-OH, and menaquinone MK-7 was the main respiratory quinone. Polar lipids contained phosphatidylethanolamine, an unidentified aminolipid, an unidentified phospholipid and other unknown lipids. Based on the data from the current polyphasic analysis, a novel species, Carboxylicivirga flava sp. -

16S Rrna Gene Metabarcoding Indicates Species-Characteristic
16S rRNA Gene Metabarcoding Indicates Species-Characteristic Microbiomes in Deep-Sea Benthic Foraminifera Iines Salonen, Panagiota-Myrsini Chronopoulou, Hidetaka Nomaki, Dewi Langlet, Masashi Tsuchiya, Karoliina Koho To cite this version: Iines Salonen, Panagiota-Myrsini Chronopoulou, Hidetaka Nomaki, Dewi Langlet, Masashi Tsuchiya, et al.. 16S rRNA Gene Metabarcoding Indicates Species-Characteristic Microbiomes in Deep- Sea Benthic Foraminifera. Frontiers in Microbiology, Frontiers Media, 2021, 12, pp.694406. 10.3389/fmicb.2021.694406. hal-03306215 HAL Id: hal-03306215 https://hal.archives-ouvertes.fr/hal-03306215 Submitted on 29 Jul 2021 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. ORIGINAL RESEARCH published: 27 July 2021 doi: 10.3389/fmicb.2021.694406 16S rRNA Gene Metabarcoding Indicates Species-Characteristic Microbiomes in Deep-Sea Benthic Foraminifera Iines S. Salonen 1,2*, Panagiota-Myrsini Chronopoulou 1, Hidetaka Nomaki 2, Dewi Langlet 2,3,4, Masashi Tsuchiya 5 and Karoliina A. Koho 1 1 Ecosystems and Environment Research Program, University -

Bacterial Taxa Based on Greengenes Database GS1A PS1B ABY1 OD1
A1: Bacterial taxa based on GreenGenes database GS1A PS1B ABY1_OD1 0.1682 0.024 Bacteria;ABY1_OD1;ABY1_OD1_unclassified 1 0 Bacteria;ABY1_OD1;FW129;FW129_unclassified 4 0 Bacteria;ABY1_OD1;FW129;KNA6-NB12;KNA6-NB12_unclassified 5 0 Bacteria;ABY1_OD1;FW129;KNA6-NB29;KNA6-NB29_unclassified 0 1 Acidobacteria 0.7907 4.509 Bacteria;Acidobacteria;Acidobacteria_unclassified 4 31 Bacteria;Acidobacteria;Acidobacteria-5;Acidobacteria-5_unclassified 0 1 Bacteria;Acidobacteria;BPC015;BPC015_unclassified 8 30 Bacteria;Acidobacteria;BPC102;BPC102_unclassified 9 43 Bacteria;Acidobacteria;Chloracidobacteria;Ellin6075;Ellin6075_unclassified 1 0 Bacteria;Acidobacteria;iii1-15;Acidobacteria-6;RB40;RB40_unclassified 0 5 Bacteria;Acidobacteria;iii1-15;iii1-15_unclassified 1 8 Bacteria;Acidobacteria;iii1-15;Riz6I;Unclassified 0 1 Bacteria;Acidobacteria;iii1-8;Unclassified 0 2 Bacteria;Acidobacteria;OS-K;OS-K_unclassified 18 17 Bacteria;Acidobacteria;RB25;RB25_unclassified 6 47 Bacteria;Acidobacteria;Solibacteres;Solibacteres_unclassified 0 1 Actinobacteria 2.1198 6.642 Bacteria;Actinobacteria;Acidimicrobidae;Acidimicrobidae_unclassified 10 70 Bacteria;Actinobacteria;Acidimicrobidae;CL500-29;ML316M-15;ML316M-15_unclassified 0 3 Bacteria;Actinobacteria;Acidimicrobidae;EB1017_group;Acidimicrobidae_bacterium_Ellin7143;Unclassified 6 1 Bacteria;Actinobacteria;Acidimicrobidae;koll13;JTB31;BD2-10;BD2-10_unclassified 1 5 Bacteria;Actinobacteria;Acidimicrobidae;koll13;JTB31;Unclassified 16 37 Bacteria;Actinobacteria;Acidimicrobidae;koll13;koll13_unclassified 81 25 Bacteria;Actinobacteria;Acidimicrobidae;Microthrixineae;Microthrixineae_unclassified -

Psychroserpens Luteus Sp. Nov., Isolated Ffrom Red Algae
Psychroserpens Luteus Sp. Nov., Isolated fFrom Red Algae Xiu-Ya Ping Shandong University Kai Wang The PLA Rocket Force Characteristic Medical Center Jin-Yu Zhang Shandong University Shu-Xin Wang Shandong University Zong-Jun Du Shandong University Da-Shuai Mu ( [email protected] ) Shandong University https://orcid.org/0000-0003-2206-9394 Research Article Keywords: Psychroserpens luteus, draft genome sequencing, novel species, prokaryotic taxonomy. Posted Date: August 26th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-836932/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/15 Abstract A Gram-stain-negative, gliding-motile, positive for catalase, facultative anaerobic, designated strain XSD401T, was isolated from the red algae of Xiaoshi Island, Shandong Province, China. Growth occurred at 20–37 °C (optimum, 33 °C), pH 5.5–9.5 (optimum, pH 6.5–7.5), and with 0.5–5% (w/v) NaCl (optimum, 3%). The main fatty acids are iso-C15:0, iso-C15:1 G, iso-C17:0 3-OH, iso-C15:0 3-OH, C16:0. Phosphatidylethanolamine (PE), three unidentied aminolipids (AL1, AL2, AL3) and one unidentied lipid (L) were the major polar lipids. The G+C content of the genomic DNA was 33.9 mol%. Strain XSD401T had the highest sequence similarity (96.88%) to the 16S rRNA gene of Psychroserpens damuponensis KCTC 23539T. The similarities with Psychroserpens burtonensis DSM 12212T was 96.31%. The dDDH values between strain XSD401T and P. damuponensis KCTC 23539T, P. burtonensis DSM 12212T, were 20.40% and 20.30%, respectively. -

Ancylomarina Psychrotolerans Sp. Nov., Isolated from Sediments of Fildes Peninsula and Emended the Description of Genus Ancylomarina
Antonie van Leeuwenhoek (2018) 111:1183–1189 https://doi.org/10.1007/s10482-018-1025-9 ORIGINAL PAPER Ancylomarina psychrotolerans sp. nov., isolated from sediments of Fildes Peninsula and emended the description of genus Ancylomarina Chao Jia . Hong-chang Cui . Yan-qiong Han . Tian-yu Fu . Rui Du . Xiao-lei Wang . Xiao-chong Shi . Xiao-Hua Zhang Received: 7 December 2017 / Accepted: 25 January 2018 / Published online: 17 February 2018 Ó Springer International Publishing AG, part of Springer Nature 2018 Abstract A Gram-stain negative, obligately anaer- of 3% (w/v) NaCl. Strain 4SWWS2-6T contained obic, non-motile, asporogenous long rod-shaped and menaquinone-7 (MK-7) as the major respiratory non-flagellated bacterial strain, designated 4SWWS2- quinone and held iso-C15:0, anteiso-C15:0 and iso- T 6 , was isolated from sediment in the intertidal zone of C15:0 3-OH as the major cellular fatty acids. The major Fildes Peninsula, Antarctica. Phylogenetic analysis polar lipids were phosphatidylethanolamine, phos- based on 16S rRNA gene sequences indicated that phatidylmonomethylethanolamine, an aminolipid, strain 4SWWS2-6T belongs to the genus Ancylo- two unidentified lipids and an unidentified phospho- marina and showed high sequence similarity with lipid. The DNA G ? C content of strain 4SWWS2-6T Ancylomarina subtilis FA102T (96.5%). Optimal was 37.6 mol%. On the basis of the polyphasic growth occurred at pH 6.5, 16 °C and in the presence analyses, strain 4SWWS2-6T is considered to repre- sent a novel species in the genus Ancylomarina, for which the name Ancylomarina psychrotolerans sp. T The GenBank Accession Number for the 16S rRNA gene nov. -

Download Download
Agr. Nat. Resour. 54 (2020) 657–664 AGRICULTURE AND NATURAL RESOURCES Journal homepage: http://anres.kasetsart.org Research article Report on microbial communities with gene functions and distribution of elements in Echinomuricea (Anthozoa: Holaxonia) from Thailand Arin Ngamniyoma,*, Thayat Sriyapaia, Wirongrong Duangjaic, Pichapak Sriyapaib a Major in Environment, Faculty of Environmental Culture and Eco-tourism, Srinakharinwirot University, Bangkok 10110, Thailand b Department of Microbiology, Faculty of Sciences, Srinakharinwirot University, Bangkok 10110, Thailand c Department of Silviculture, Faculty of Forestry, Kasetsart University, Bangkok 10900, Thailand Article Info Abstract Article history: The genus Echinomuricea consists of marine invertebrates that are important to ocean ecosystems. Received 23 March 2020 The present study provided the first data on the microsymbiont community with microbial gene Revised 17 June 2020 Accepted 28 June 2020 profiles and depositions of elements in gorgonian Echinomuricea cf. pulchra from parts of the Available online 30 December 2020 western Gulf of Thailand. Among all the microbes identified, the bacterial diversity was the most abundant in gorgonian corals. The Vibrionaceae and Marinilabiliaceae were the predominant Keywords: identified Gammaproteobacteria and Bacteroidetes, respectively. Of the other microbes, the Chemical deposition, Echinomuricea were dominated by the Mucorales for fungi, the Nitrosopumilales for Archaea and Gene prediction, Gorgonians, the Herpesvirales for viruses. In -

Labilibacter Aurantiacus Gen. Nov., Sp. Nov., Isolated from Sea Squirt (Styela Clava) and Reclassification of Saccharicrinis Marinus As Labilibacter Marinus Comb
NOTE Lu et al., Int J Syst Evol Microbiol 2017;67:441–446 DOI 10.1099/ijsem.0.001649 Labilibacter aurantiacus gen. nov., sp. nov., isolated from sea squirt (Styela clava) and reclassification of Saccharicrinis marinus as Labilibacter marinus comb. nov. De-Chen Lu,1 Jin-Xin Zhao,1 Feng-Qing Wang,1 Zhi-Hong Xie2,* and Zong-Jun Du1,* Abstract A Gram-stain-negative, facultatively anaerobic, orange-pigmented bacterium, designated HQYD1T, was isolated from a sea squirt (Styelaclava) and characterized using a polyphasic approach. Morphologically, strain HQYD1T exhibited rods with gliding motility. This novel isolate grew optimally at 28 C in the presence of 2–3 % (w/v) NaCl. The 16S rRNA gene sequence was most similar to [Saccharicrinis] marinus Y11T (96.3 %), followed by Saccharicinis fermentans DSM 9555T (93.8 %). The T dominant fatty acids of strain HQYD1 were identified as C16 : 0,C18 : 0 and iso-C15 : 0. Major polar lipids included an unidentified lipid and a phospholipid. The major respiratory quinone was found to be MK-7, and the genomic DNA G+C content was determined to be 35.1 mol%. Based on evidence from this taxonomic study, a novel genus, Labilibacter gen. nov., is proposed in the family Marinilabiliaceae with type species Labilibacter aurantiacus sp. nov. The type strain of the type species is HQYD1T (=MCCC 1K02304T=KCTC 42583T). As [Saccharicrinis] marinus Y11T clustered phylogenetically with strain HQYD1T, we also propose [Saccharicrinis] marinus Y11T be reclassified as Labilibacter marinus comb. nov. (type strain Y11T=CICC 10837T=KCTC 42400T). The family Marinilabiliaceae suggested by Ludwig et al. -

16S Rrna Gene Metabarcoding Indicates Species-Characteristic Microbiomes in Deep-Sea Benthic Foraminifera
ORIGINAL RESEARCH published: 27 July 2021 doi: 10.3389/fmicb.2021.694406 16S rRNA Gene Metabarcoding Indicates Species-Characteristic Microbiomes in Deep-Sea Benthic Foraminifera Iines S. Salonen 1,2*, Panagiota-Myrsini Chronopoulou 1, Hidetaka Nomaki 2, Dewi Langlet 2,3,4, Masashi Tsuchiya 5 and Karoliina A. Koho 1 1 Ecosystems and Environment Research Program, University of Helsinki, Helsinki, Finland, 2 SUGAR, X-star, Japan Agency of Marine-Earth Science and Technology (JAMSTEC), Yokosuka, Japan, 3 UMR 8187 - LOG - Laboratoire d’Océanologie et de Géosciences, Université de Lille - CNRS, Université du Littoral Côte d’Opale, Station Marine de Wimereux, Lille, France, 4 Evolution, Cell Biology, and Symbiosis Unit, Okinawa Institute of Science and Technology, Okinawa, Japan, 5 Research Institute for Global Change (RIGC), Japan Agency of Marine-Earth Science and Technology (JAMSTEC), Yokosuka, Japan Foraminifera are unicellular eukaryotes that are an integral part of benthic fauna in many marine ecosystems, including the deep sea, with direct impacts on benthic biogeochemical cycles. In these systems, different foraminiferal species are known to have a distinct vertical Edited by: distribution, i.e., microhabitat preference, which is tightly linked to the physico-chemical Francisco J. A. Nascimento, Stockholm University, Sweden zonation of the sediment. Hence, foraminifera are well-adapted to thrive in various conditions, Reviewed by: even under anoxia. However, despite the ecological and biogeochemical significance of Amir Szitenberg, foraminifera, their ecology remains poorly understood. This is especially true in terms of the Dead Sea and Arava Science Center – Dead Sea Branch, Israel composition and diversity of their microbiome, although foraminifera are known to harbor Fatma Gomaa, diverse endobionts, which may have a significant meaning to each species’ survival strategy. -

Tangfeifania Diversioriginum Gen. Nov., Sp. Nov., a Representative of the Family Draconibacteriaceae
International Journal of Systematic and Evolutionary Microbiology (2014), 64, 3473–3477 DOI 10.1099/ijs.0.066902-0 Tangfeifania diversioriginum gen. nov., sp. nov., a representative of the family Draconibacteriaceae Qian-Qian Liu,1 Xiao-Li Li,1 Alejandro P. Rooney,2 Zong-Jun Du1,3 and Guan-Jun Chen1,3 Correspondence 1College of Marine Science, Shandong University at Weihai, Weihai 264209, PR China Zong-Jun Du 2National Center for Agricultural Utilization Research, Agricultural Research Service, [email protected] US Department of Agriculture, Peoria, IL 61604, USA 3State key Laboratory of Microbial Technology, Shandong University, Jinan 250100, PR China A novel Gram-stain-negative, facultatively anaerobic, catalase- and oxidase-positive, non-motile and pink-pigmented bacterium, designated G22T, was isolated from Gahai, a saltwater lake in Qinghai province, China. Optimal growth occurred at 33–35 6C, pH 7.0–7.5, and in the presence of 2–4 % (w/v) NaCl. The DNA G+C content was 40.0 mol%. The major polar lipids were phosphatidylethanolamine and three unknown lipids. The predominant cellular fatty acids were iso-C15 : 0, anteiso-C15 : 0, iso-C17 : 0 3-OH and iso-C15 : 0 3-OH, and MK-7 was the main respiratory quinone. Phylogenetic analysis based on 16S rRNA gene sequences revealed that strain G22T fell within the class Bacteroidia. Its closest phylogenetic neighbour was the recently described species Draconibacterium orientale, the sole member of the family Draconibacteriaceae, with merely 90.04 % sequence similarity. On the basis of phenotypic, chemotaxonomic and phylogenetic evidence observed, a novel species in a new genus, Tangfeifania diversioriginum gen. -

Mariniphaga Sediminis Sp. Nov., Isolated from Coastal Sediment Feng-Qing Wang,1 Qi-Yao Shen,1 Guan-Jun Chen1,2 and Zong-Jun Du1,2
International Journal of Systematic and Evolutionary Microbiology (2015), 65, 2908–2912 DOI 10.1099/ijs.0.000354 Mariniphaga sediminis sp. nov., isolated from coastal sediment Feng-Qing Wang,1 Qi-Yao Shen,1 Guan-Jun Chen1,2 and Zong-Jun Du1,2 Correspondence 1College of Marine Science, Shandong University at Weihai, Weihai 264209, PR China Zong-Jun Du 2State Key Laboratory of Microbial Technology, Shandong University, Jinan 250100, PR China [email protected] A Gram-stain-negative and facultatively anaerobic bacterium, SY21T, was isolated from marine sediments of the coastal area in Weihai, China (1228 09 370 E378 319 330 N). Cells of strain SY21T were 0.3–0.5 mm wide and 1.5–2.5 mm long, catalase- and oxidase-positive. Colonies on 2216E agar were transparent, beige- to pale-brown-pigmented, and approximately 0.5 mm in diameter. Growth occurred optimally at 33–37 8C, pH 7.0–7.5 and in the presence of 2–3 % (w/v) NaCl. Phylogenetic analysis of the 16S rRNA gene indicated that strain SY21T was a member of the genus Mariniphaga within the family Prolixibacteraceae. The closest described neighbour in terms of 16S rRNA gene sequences identity was Mariniphaga anaerophila Fu11-5T (94.7 %). The major respiratory quinone of strain SY21T was MK-7, and the dominant fatty acids were iso-C15 : 0, iso-C17 : 0 3-OH and anteiso-C15 : 0.The major polar lipids were phosphatidylethanolamine, aminolipid and an unidentified lipid, and the DNA G+C content was 37.9 mol%. The distinct phylogenetic position and phenotypic traits distinguished the novel isolate from M. -
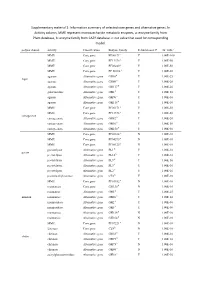
Information Summary of Selected Core Genes and Alternative Genes. in Activity Column, MME Represent Mo
Supplementary material 1: Information summary of selected core genes and alternative genes. In Activity column, MME represent monosaccharide metabolic enzymes, a: enzyme family from Pfam database, b: enzyme family from CAZY database. c: cut value that used for corresponding model. polysaccharide activity Classification Enzyme family Rebuild model? cut_valuec MME Core gene PF00171a Y 1.00E-100 MME Core gene PF13378 a Y 1.00E-50 MME Core gene PF08240 a Y 1.00E-50 MME Core gene PF 00106 a Y 1.00E-50 agarase Alternative gene GH16b Y 1.00E-25 Agar agarase Alternative gene GH86 b Y 1.00E-20 agarase Alternative gene GH117 b Y 1.00E-20 galactosidase Alternative gene GH2 b Y 1.00E-50 agarase Alternative gene GH96 b Y 1.00E-10 agarase Alternative gene GH118 b Y 1.00E-10 MME Core gene PF00171 a Y 1.00E-50 MME Core gene PF13378 a Y 1.00E-50 carrageenan carrageenase Alternative gene GH82 b Y 1.00E-20 carrageenase Alternative gene GH16 b Y 1.00E-50 carrageenase Alternative gene GH150 b Y 1.00E-50 MME Core gene PF02614 a N 1.00E-10 MME Core gene PF04295 a N 1.00E-10 MME Core gene PF08125 a N 1.00E-10 pectatelyase Alternative gene PL1 b Y 1.00E-10 pectin pectatelyase Alternative gene PL10 b Y 1.00E-10 pectatelyase Alternative gene PL9 b Y 1.00E-50 pectatelyase Alternative gene PL3 b Y 1.00E-10 pectatelyase Alternative gene PL2 b Y 1.00E-10 pectinmethylesterase Alternative gene CE8 b Y 1.00E-10 MME Core gene PF01182 a N 1.00E-10 mannanase Core gene GH130 b N 1.00E-10 mannanase Alternative gene GH5 b Y 1.00E-25 mannan mannanase Alternative gene GH26 b