Some Reminiscences of Mass Spectrometry and the Manhattan Project
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Taming of “49” Big Science in Little Time
The Taming of “49” Big science in little time Recollections of Edward F. Hammel During the Manhattan Project, plutonium was often referred to, simply, as 49. Number 4 was for the last digit in 94 (the atomic number of plutonium) and 9 for the last digit in plutonium-239, the isotope of choice for nuclear weapons. The story that unfolds was adapted from Plutonium Metallurgy at Los Alamos, 1943–1945, as Edward F. Hammel remembers the events of those years. 48 Los Alamos Science Number 26 2000 The Taming of “49” he work in plutonium chemistry tion work was an inevitable conse- the metal could be fabricated into and metallurgy carried out at quence of the nuclear and physical satisfactory weapon components. TLos Alamos (Site Y) between research that was still to be conducted In addition, not until January 1944 1943 and 1945 had a somewhat contro- on the metal. It would clearly have did the first few milligrams of pile- versial history. The controversy was been inefficient and time consuming to produced plutonium arrive at Los about who was going to do what. ship small amounts of plutonium metal Alamos. The first 1-gram shipment At the time Los Alamos was being back to Chicago for repurification and arrived in February 1944, and quantity organized, most of the expertise in plu- refabrication into different sizes and shipments of plutonium did not begin to tonium chemistry resided at Berkeley, shapes for the next-scheduled nuclear arrive at Los Alamos until May 1945. where plutonium was discovered in physics experiment. From the outset, it was clear that the December 1940, and at the Met Lab in Minimizing the time spent to solve purification of plutonium was the most Chicago. -
Laser Isotope Separation (LIS), Technical and Economic
NASA TECHNICAL MEMORANDUM A STATUS OF PROGRESS FOR THL LASER lsofopE SEPARATION (11 SI PROCESS +tear 1976 NASA George C. Mdr~bdlSpace Flight Center Marshdl Space Fb$t Center, Alabama lLSFC - Form 3190 (Rev June 1971) REPORT STANDARD TITLE PACE I nEPMTn0. 3. RECIPIENT*$ CATILOC NO. NASA TM X-73345 10 TITLE UO SUTlTLt IS. REPORT DATE I September A st.tUaof for Iaser isotOpe ¶tian lS76 I Progress the (LIS) 6 PERFWYIIIG WGUIZATIO* CQOE George C. M8ralmll!3gam Flight Center I 1. COUTRUT OR am yo. I MarW Flight Center, Alabama 35812 Tecbnid Memormdum National Aemutics and Space Administration Washingtan, D.C. 20546 I I Prepared by Systems Aaalysis and Integration Iaboratory, Science and Engineering An overview of the various categories of the LE3 methodology is given together with illustrations showing a simplified version of the LIS tecbnique, an example of the two-phoiin photoionization category, and a diagram depicting how the energy levels of various isdope influence the LIS process. A&icatlons have been proposed for the LIS system which, in addition to the use to enrich uranium, could in themselves develop into programs of tremendous scope and breadth. Such applications as treatment of radioac '--ewastes from light-water nKzlear reactors, enriching the deuterlum isotope to make heavv-water, and enrlchhg tik light isotopes of such 17 KEt WORDS 18. DISTRIBUTION STATEMENT 5ECUQlTY CLASSIF. Ff thh PI*) 21 NO. OF PAbFS 22 PRICE Unclassified Unclassified I 20 NTIS PREFACE Since the publication of t& first Techid hiemomxitun (TM X-64947) on the Laser hotope Separation (LE)process in May 1975 [l], there bbeen a virtual explosion of available information on this process. -

Extensive Interest in Nuclear Fuel Cycle Technologies
Institute for Science and International Security ISIS REPORT March 19, 2012 Department 70 and the Physics Research Center: Extensive Interest in Nuclear Fuel Cycle Technologies By David Albright, Paul Brannan, Mark Gorwitz, and Andrew Ortendahl On February 23, 2012, ISIS released the report, The Physics Research Center and Iran’s Parallel Military Nuclear Program, in which ISIS evaluated a set of 1,600 telexes outlining a set of departments or buying centers of the former Physics Research Center (PHRC). These departments appeared to be purchasing a variety of goods for specific nuclear technologies, including gas centrifuges, uranium conversion, uranium exploration and perhaps mining, and heavy water production. Figure 1 is a list of the purposes of these departments. The telexes are evaluated in more depth in the February 23, 2012 ISIS report and support that, contrary to Iran’s statements to the International Atomic Energy Agency (IAEA), the PHRC ran a parallel military nuclear program in the 1990s. In the telexes, ISIS identified a department called Department 70 that is linked to the PHRC. This department tried to procure or obtained technical publications and reports from a document center, relevant know-how from suppliers, catalogues from suppliers about particular goods, and a mini- computer from the Digital Equipment Corporation. Department 70 appears to have had personnel highly knowledgeable about the existing literature on a variety of fuel cycle technologies, particularly gas centrifuges. Orders to a British document center reveal many technical publications about gas centrifuges, atomic laser isotope enrichment, the production of uranium compounds including uranium tetrafluoride and uranium hexafluoride (and precursors such as hydrofluoric acid), nuclear grade graphite, and the production of heavy water. -

Secrets Jeremy Bernstein
INFERENCE / Vol. 6, No. 1 Secrets Jeremy Bernstein Restricted Data: The History of Nuclear Secrecy in the decided to found a rival weapons laboratory. Even if Teller United States had offered me a job, I doubt that I would have accepted.3 by Alex Wellerstein After obtaining my degree, I was offered a job that University of Chicago Press, 528 pp., $35.00. would keep me in Cambridge for at least another year. One year became two and at the end of my second year I was uclear weapons have been shrouded in secrecy accepted at the Institute for Advanced Study in Princeton. from the very beginning. After plutonium was It was around this time that the chairman of the physics discovered at the University of California in department at Harvard, Kenneth Bainbridge, came to me NDecember 1940, researchers led by Glenn Seaborg submit- with an offer. Bainbridge had been an important figure at ted a pair of letters to the Physical Review. The details of Los Alamos during the war. Robert Oppenheimer had put their discovery were withheld from publication until after him in charge of the site in New Mexico where the Trinity the war.1 Once the project to make a nuclear weapon got test had taken place.4 Bainbridge told me that the labora- underway, secrecy became a very serious matter indeed. tory was offering summer jobs to young PhDs and asked The story of these efforts and how they evolved after the if I was interested. I was very interested. Los Alamos had war is the subject of Alex Wellerstein’s Restricted Data: an almost mystical significance for me due to its history The History of Nuclear Secrecy in the United States. -

Cold War Requisitions, Scientific Manpower, and the Production of American Physicists After World War II
DAVID KAISER* Cold War requisitions, scientific manpower, and the production of American physicists after World War II 1. RAYMOND BIRGE’S “MAIN OBJECTIVE” “THE MAIN OBJECTIVE of this department of physics,” Raymond Birge wrote in late May 1955, “is to train Ph.D.’s in physics.” Birge— iconic, somber, a displaced Yankee who traced his New England ancestry nine generations back—had been chair of Berkeley’s physics department for twenty-two years; by the mid-1950s, it was the nation’s largest. At the time he explained his department’s “main objec- tive,” Birge was the retiring president of the American Physical Society (APS). Birge and his colleagues in Berkeley’s physics department had emphasized the importance of its graduate program many times before in annual budget requests to the university administration and in funding reports to private industries; it would be easy to read such remarks as thinly-veiled requests for more funding, since training physics Ph.D.s became expensive after World War II. This time, however, Birge articulated his department’s mission in a letter to a local citizen, far outside of the university bureaucracy, who had no funds to offer and who had requested no such pronouncement. 1 *Program in Science, Technology, and Society, and Department of Physics, Building E51- 185, MIT, Cambridge, MA 02139; [email protected]. My thanks to Shane Hamilton for his research assistance, and to Alexis De Greiff, Kenji Ito, John Krige, Elizabeth Paris, and John Rudolph for their helpful comments on an earlier draft. The following abbreviations are used: AIP-EMD, American Institute of Physics, Edu- cation and Manpower Division Records, Niels Bohr Library, American Institute of Physics, College Park, MD; BAS, Bulletin of the atomic scientists; BDP , University of California, Berkeley, Department of Physics Records, Bancroft Library, Berkeley, CA; HDP, Harvard University Department of Physics Records, Pusey Library, Cambridge, MA; PDP, Princeton University Department of Physics Records, Seeley G. -

THE MEETING Meridel Rubenstein 1995
THE MEETING Meridel Rubenstein 1995 Palladium prints, steel, single-channel video Video assistance by Steina Video run time 4:00 minutes Tia Collection The Meeting consists of twenty portraits of people from San Ildefonso Pueblo and Manhattan Project physicists—who met at the home of Edith Warner during the making of the first atomic bomb—and twenty photographs of carefully selected objects of significance to each group. In this grouping are people from San Ildefonso Pueblo and the objects they selected from the collections of the Museum of Indian Arts and Culture to represent their culture. 1A ROSE HUGHES 2A TALL-NECKED JAR 3A BLUE CORN 4A SLEIGH BELLS 5A FLORENCE NARANJO Rose Hughes holding a photograph of WITH AVANYU One of the most accomplished and (Museum of Indian Arts and Culture) Married to Louis Naranjo; her father, Tony Peña, who organized (plumed serpent) made by Julian and recognized of the San Ildefonso Sleigh bells are commonly used in granddaughter of Ignacio and Susana the building of Edith Warner’s second Maria Martinez, ca. 1930 (Museum of potters. Like many women from the ceremonial dances to attract rain. Aguilar; daughter of Joe Aguilar, who house. Hughes worked at Edith Indian Arts and Culture) Edith Warner pueblos, she worked as a maid for the Tilano Montoya returned with bells like helped Edith Warner remodel the Warner’s with Florence Naranjo one was shown a pot like this one in 1922 Oppenheimers. these from Europe, where he went on tearoom. Edith called her Florencita. summer. She recalls that Edith once on her first visit to San Ildefonso, in the tour with a group of Pueblo dancers. -

Development of a Viable Route for Lithium-6 Supply of DEMO and Future Fusion Power Plants T
Fusion Engineering and Design 149 (2019) 111339 Contents lists available at ScienceDirect Fusion Engineering and Design journal homepage: www.elsevier.com/locate/fusengdes Development of a viable route for lithium-6 supply of DEMO and future fusion power plants T T. Giegerich⁎, K. Battes, J.C. Schwenzer, C. Day Karlsruhe Institute of Technology (KIT), Hermann-von-Helmholtz-Platz 1, 76344 Eggenstein-Leopoldshafen, Germany ARTICLE INFO ABSTRACT Keywords: In the European DEMO program, the design development of a demonstration power plant (DEMO) is currently in DEMO its pre-conceptual phase. In DEMO, breeding blankets will use large quantities of lithium, enriched in the isotope Lithium-6 lithium-6 (6Li), for breeding the tritium needed to feed the DT fusion reaction. Unfortunately, enriched lithium is Lithium enrichment commercially not available in the required quantities, which is threatening the success of future power plant ICOMAX process applications of nuclear fusion. Even if the manufacturing of the breeding blankets is still two decades ahead of Mercury us, it is now mandatory to address the topic of lithium-6 supply and to make sure that a viable supply (and HgLab reprocessing) route is available when needed. This paper presents an unbiased systems engineering approach assessing a number of available lithium iso- tope separation methods by defining requirements, rating them systematically and finally calculating a ranking number expressing the value of different methods. As a result, we suggest using a chemical exchange method based on a lithium amalgam system, but including some important improvements leading to a more efficient and ‘clean’ process (the ICOMAX process) in comparison with the formerly used COLEX process. -

Trinity Site July 16, 1945
Trinity Site July 16, 1945 "The effects could well be called unprecedented, magnificent, beauti ful, stupendous, and terrifying. No man-made phenomenon of such tremendous power had ever occurred before. The lighting effects beggared description. The whole country was lighted by a searing light with the intensity many times that of the midday sun." Brig. Gen. Thomas Farrell A national historic landmark on White Sands Missile Range -- www.wsmr.army.mil Radiation Basics Radiation comes from the nucJeus of the gamma ray. This is a type of electromag individual atoms. Simple atoms like oxygen netic radiation like visible light, radio waves are very stable. Its nucleus has eight protons and X-rays. They travel at the speed of light. and eight neutrons and holds together well. It takes at least an inch of lead or eight The nucJeus of a complex atom like inches of concrete to stop them. uranium is not as stable. Uranium has 92 Finally, neutrons are also emitted by protons and 146 neutrons in its core. These some radioactive substances. Neutrons are unstable atoms tend to break down into very penetrating but are not as common in more stable, simpler forms. When this nature. Neutrons have the capability of happens the atom emits subatomic particles striking the nucleus of another atom and and gamma rays. This is where the word changing a stable atom into an unstable, and "radiation" comes from -- the atom radiates therefore, radioactive one. Neutrons emitted particles and rays. in nuc!ear reactors are contained in the Health physicists are concerned with reactor vessel or shielding and cause the four emissions from the nucleus of these vessel walls to become radioactive. -
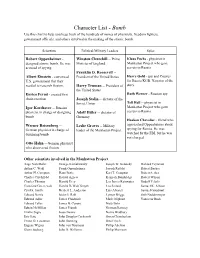
Character List
Character List - Bomb Use this chart to help you keep track of the hundreds of names of physicists, freedom fighters, government officials, and others involved in the making of the atomic bomb. Scientists Political/Military Leaders Spies Robert Oppenheimer - Winston Churchill -- Prime Klaus Fuchs - physicist in designed atomic bomb. He was Minister of England Manhattan Project who gave accused of spying. secrets to Russia Franklin D. Roosevelt -- Albert Einstein - convinced President of the United States Harry Gold - spy and Courier U.S. government that they for Russia KGB. Narrator of the needed to research fission. Harry Truman -- President of story the United States Enrico Fermi - created first Ruth Werner - Russian spy chain reaction Joseph Stalin -- dictator of the Tell Hall -- physicist in Soviet Union Igor Korchatov -- Russian Manhattan Project who gave physicist in charge of designing Adolf Hitler -- dictator of secrets to Russia bomb Germany Haakon Chevalier - friend who Werner Reisenberg -- Leslie Groves -- Military approached Oppenheimer about German physicist in charge of leader of the Manhattan Project spying for Russia. He was designing bomb watched by the FBI, but he was not charged. Otto Hahn -- German physicist who discovered fission Other scientists involved in the Manhattan Project: Aage Niels Bohr George Kistiakowsky Joseph W. Kennedy Richard Feynman Arthur C. Wahl Frank Oppenheimer Joseph Rotblat Robert Bacher Arthur H. Compton Hans Bethe Karl T. Compton Robert Serber Charles Critchfield Harold Agnew Kenneth Bainbridge Robert Wilson Charles Thomas Harold Urey Leo James Rainwater Rudolf Pelerls Crawford Greenewalt Harold DeWolf Smyth Leo Szilard Samuel K. Allison Cyril S. Smith Herbert L. Anderson Luis Alvarez Samuel Goudsmit Edward Norris Isidor I. -

Atomic Energy Je^K L'energie Atomique of Canada Umited Ksv Du Canada Li Mite E
AECL-7416 ATOMIC ENERGY JE^K L'ENERGIE ATOMIQUE OF CANADA UMITED KSV DU CANADA LI MITE E PROGRESS REPORT CHEMISTRY AND MATERIALS DIVISION 1 April -30 June, 1981 PR-CMa-57 Chalk River Nuclear Laboratories Chalk River, Ontario August 1981 PREVIOUS REPORTS IN THIS SERIES PR-CMa-56 January 1 to March 31, 1981 AECL-7332 PR-CMa-55 October 1 to December 31, 1980 AECL-7242 PR-CHa-54 July 1 to September 30, 1980 AECL-7156 PR-CMa-53 April 1 to June 30, 1980 AECL-7094 PR-CMa-57 ATOMIC ENERGY OF CANADA LIMITED PROGRESS REPORT April 1, 1981 - June 30, 1981 CHEMISTRY AND MATERIALS DIVISION The results and conclusions given here are not classified or restricted in any way; however, some of the information is of a preliminary nature. Readers interested in using the information in their own research are invited to consult with the contributors for further details. Copies of the AECL publications referred to in this report may be obtained by writing to the Scientific Document Distribution Offfee, Chalk River Nuclear Laboratories, Chalk River, Ontario, KOJ 1J0. Chalk River Nuclear Laboratories Chalk River, Ontario 1981 August AECL-7416 PROGRESS REPORT April 1, 1981 - June 30, 1981 CHEMISTRY AND MATERIALS DIVISION Director - T. A. Eastwood Secretary - Ms. A. E. Goodale Contents Page HIGHLIGHTS T.A. Eastwood (i) 1. SOLID STATE SCIENCE BRANCH I.V. Mitchell 1 2. GENERAL CHEMISTRY BRANCH I.H. Crocker 27 3. PHYSICAL CHEMISTRY BRANCH D.R. Smith 53 4. MATERIALS SCIENCE BRANCH B. Cox 71 (i) CHEMISTRY AND MATERIALS DIVISION HIGHLIGHTS T. -

Lithium Isotope Separation a Review of Possible Techniques
Canadian Fusion Fuels Technology Project Lithium Isotope Separation A Review of Possible Techniques Authors E.A. Symons Atomic Energy of Canada Limited CFFTP GENERAL CFFTP Report Number s Cross R< Report Number February 1985 CFFTP-G-85036 f AECL-8 The Canadian Fusion Fuels Technology Project represents part of Canada's overall effort in fusion development. The focus for CFFTP is tritium and tritium technology. The project is funded by the governments of Canada and Ontario, and by Ontario Hydro. The Project is managed by Ontario Hydro. CFFTP will sponsor research, development and studies to extend existing experience and capability gained in handling tritium as part of the CANDU fission program. It is planned that this work will be in full collaboration and serve the needs of international fusion programs. LITHIUM ISOTOPE SEPARATION: A REVIEW OF POSSIBLE TECHNIQUES Report No. CFFTP-G-85036 Cross Ref. Report No. AECL-8708 February, 1985 by E.A. Symons CFFTP GENERAL •C - Copyright Ontario Hydro, Canada - (1985) Enquiries about Copyright and reproduction should be addressed to: Program Manager, CFFTP 2700 Lakeshore Road West Mississauga, Ontario L5J 1K3 LITHIUM ISOTOPE SEPARATION: A REVIEW OF POSSIBLE TECHNIQUES Report No. OFFTP-G-85036 Cross-Ref Report No. AECL-87O8 February, 1985 by E.A. Symons (x. Prepared by: 0 E.A. Symons Physical Chemistry Branch Chemistry and Materials Division Atomic Energy of Canada Limited Reviewed by: D.P. Dautovich Manager - Technology Development Canadian Fusion Fuels Technology Project Approved by: T.S. Drolet Project Manager Canadian Fusion Fuels Technology Project ii ACKNOWLEDGEMENT This report has been prepared under Element 4 (Lithium Compound Chemistry) of the Fusion Breeder Blanket Program, which is jointly funded by AECL and CFFTP. -

Uranium Enrichment Plant Characteristics—A Training Manual for the IAEA
ORNL/TM-2019/1311 ISPO-553 Uranium Enrichment Plant Characteristics—A Training Manual for the IAEA J. M. Whitaker October 2019 Approved for public release. Distribution is unlimited. DOCUMENT AVAILABILITY Reports produced after January 1, 1996, are generally available free via US Department of Energy (DOE) SciTech Connect. Website www.osti.gov Reports produced before January 1, 1996, may be purchased by members of the public from the following source: National Technical Information Service 5285 Port Royal Road Springfield, VA 22161 Telephone 703-605-6000 (1-800-553-6847) TDD 703-487-4639 Fax 703-605-6900 E-mail [email protected] Website http://classic.ntis.gov/ Reports are available to DOE employees, DOE contractors, Energy Technology Data Exchange representatives, and International Nuclear Information System representatives from the following source: Office of Scientific and Technical Information PO Box 62 Oak Ridge, TN 37831 Telephone 865-576-8401 Fax 865-576-5728 E-mail [email protected] Website http://www.osti.gov/contact.html This report was prepared as an account of work sponsored by an agency of the United States Government. Neither the United States Government nor any agency thereof, nor any of their employees, makes any warranty, express or implied, or assumes any legal liability or responsibility for the accuracy, completeness, or usefulness of any information, apparatus, product, or process disclosed, or represents that its use would not infringe privately owned rights. Reference herein to any specific commercial product, process, or service by trade name, trademark, manufacturer, or otherwise, does not necessarily constitute or imply its endorsement, recommendation, or favoring by the United States Government or any agency thereof.