The Reactivity and Selectivity of the Reaction of Sulfur Trioxide and Bromobenzene
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bromobenzene D5
Safety data sheet according to Regulation (EC) No. 1907/2006 (REACH), amended by 2015/830/EU Bromobenzene D5 99,5 Atom%D article number: HN93 date of compilation: 2020-09-02 Version: 1.0 en SECTION 1: Identification of the substance/mixture and of the company/ undertaking 1.1 Product identifier Identification of the substance Bromobenzene D5 99,5 Atom%D Article number HN93 Registration number (REACH) It is not required to list the identified uses be- cause the substance is not subject to registration according to REACH (< 1 t/a) EC number 224-013-8 CAS number 4165-57-5 1.2 Relevant identified uses of the substance or mixture and uses advised against Identified uses: laboratory chemical laboratory and analytical use 1.3 Details of the supplier of the safety data sheet Carl Roth GmbH + Co KG Schoemperlenstr. 3-5 D-76185 Karlsruhe Germany Telephone: +49 (0) 721 - 56 06 0 Telefax: +49 (0) 721 - 56 06 149 e-mail: [email protected] Website: www.carlroth.de Competent person responsible for the safety data : Department Health, Safety and Environment sheet: e-mail (competent person): [email protected] 1.4 Emergency telephone number Name Street Postal code/ Telephone Website city National Poisons Inform- Dudley Rd B187QH Birm- 844 892 0111 ation Service ingham City Hospital Emergency information service +49/(0)89 19240 SECTION 2: Hazards identification 2.1 Classification of the substance or mixture Classification according to Regulation (EC) No 1272/2008 (CLP) Classification acc. to GHS Section Hazard class Hazard class and cat- Hazard egory state- ment 2.6 flammable liquid (Flam. -

Chem 353: Grignard
GRIG.1 ORGANIC SYNTHESIS: BENZOIC ACID VIA A GRIGNARD REACTION TECHNIQUES REQUIRED : Reflux with addition apparatus, rotary evaporation OTHER DOCUMENTS Experimental procedure, product spectra INTRODUCTION In this experiment you will synthesise benzoic acid using bromobenzene to prepare a Grignard reagent, which is then reacted with carbon dioxide, worked-up and purified to give the acid. This sequence serves to illustrate some important concepts of practical synthetic organic chemistry : preparing and working with air and moisture sensitive reagents, the "work-up", extractions, apparatus set-up, etc. The synthesis utilises one of the most important type of reagents discussed in introductory organic chemistry, organometallic reagents. In this reaction, the Grignard reagent (an organomagnesium compound), phenylmagnesium bromide is prepared by reaction of bromobenzene with magnesium metal in diethyl ether (the solvent). The Grignard reagent will then be converted to benzoic acid via the reaction of the Grignard reagent with excess dry ice (solid CO2) followed by a "work-up" using dilute aqueous acid : The aryl (or alkyl) group of the Grignard reagent behaves as if it has the characteristics of a carbanion so it is a source of nucleophilic carbon. It is reasonable to represent the structure of the - + Grignard reagent as a partly ionic compound, R ....MgX. This partially-bonded carbanion is a very strong base and will react with acids (HA) to give an alkane: RH + MgAX RMgX + HA Any compound with suitably acidic hydrogens will readily donate a proton to destroy the reagent. Water, alcohols, terminal acetylenes, phenols and carboxylic acids are just some of the functional groups that are sufficiently acidic to bring about this reaction which is usually an unwanted side reaction that destroys the Grignard reagent. -

TOXICOLOGICAL REVIEW of BROMOBENZENE (CAS No
EPA/635/R-07/002F www.epa.gov/iris TOXICOLOGICAL REVIEW OF BROMOBENZENE (CAS No. 108-86-1) In Support of Summary Information on the Integrated Risk Information System (IRIS) September 2009 U.S. Environmental Protection Agency Washington, DC DISCLAIMER This document has been reviewed in accordance with U.S. Environmental Protection Agency policy and approved for publication. Mention of trade names or commercial products does not constitute endorsement or recommendation for use. ii CONTENTS−TOXICOLOGICAL REVIEW OF BROMOBENZENE (CAS No. 108-86-1) LIST OF TABLES......................................................................................................................... vi LIST OF FIGURES ....................................................................................................................... ix LIST OF ABBREVIATIONS AND ACRONYMS ....................................................................... x FOREWORD ................................................................................................................................. xi AUTHORS, CONTRIBUTORS, AND REVIEWERS ................................................................ xii 1. INTRODUCTION ..................................................................................................................... 1 2. CHEMICAL AND PHYSICAL INFORMATION RELEVANT TO ASSESSMENTS .......... 3 3. TOXICOKINETICS .................................................................................................................. 6 3.1. ABSORPTION ................................................................................................................ -

The Ozonolysis of Phenyl Grignard Reagent
University of Montana ScholarWorks at University of Montana Graduate Student Theses, Dissertations, & Professional Papers Graduate School 1971 The ozonolysis of phenyl Grignard reagent Gale Manning Sherrodd The University of Montana Follow this and additional works at: https://scholarworks.umt.edu/etd Let us know how access to this document benefits ou.y Recommended Citation Sherrodd, Gale Manning, "The ozonolysis of phenyl Grignard reagent" (1971). Graduate Student Theses, Dissertations, & Professional Papers. 8297. https://scholarworks.umt.edu/etd/8297 This Thesis is brought to you for free and open access by the Graduate School at ScholarWorks at University of Montana. It has been accepted for inclusion in Graduate Student Theses, Dissertations, & Professional Papers by an authorized administrator of ScholarWorks at University of Montana. For more information, please contact [email protected]. THE OZONOLYSIS OF PHENYL GRIGNARD REAGENT By Gale M. Sherrodd B.S., Rocky Mountain College, I969 Presented in partial fulfillment of the requirements for the degree of Master of Arts for Teachers UNIVERSITY OF MONTANA 1971 Approved by: Chairman, Board of Examiners De^ , Graduate *School / n ? / Date Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. UMI Number: EP39098 All rights reserved INFORMATION TO ALL USERS The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed, a note will indicate the deletion. UMT DiMMtstion PuWiahing UMI EP39098 Published by ProQuest LLC (2013). Copyright in the Dissertation held by the Author. -

TOXICOLOGICAL REVIEW of BROMOBENZENE (CAS No
DRAFT - DO NOT CITE OR QUOTE EPA/635/R-07/002 www.epa.gov/iris TOXICOLOGICAL REVIEW OF BROMOBENZENE (CAS No. 108-86-1) In Support of Summary Information on the Integrated Risk Information System (IRIS) June 2007 NOTICE This document is an interagency review draft. It has not been formally released by the U.S. Environmental Protection Agency and should not at this stage be construed to represent Agency position on this chemical. It is being circulated for review of its technical accuracy and science policy implications. U.S. Environmental Protection Agency Washington, DC DISCLAIMER This document is a preliminary draft for review purposes only and does not constitute U.S. Environmental Protection Agency policy. Mention of trade names or commercial products does not constitute endorsement or recommendation for use. 6/7/07 ii DRAFT – DO NOT CITE OR QUOTE CONTENTSCTOXICOLOGICAL REVIEW OF BROMOBENZENE (CAS No. 108-86-1) LIST OF FIGURES ....................................................................................................................... vi LIST OF TABLES........................................................................................................................ vii LIST OF ABBREVIATIONS AND ACRONYMS ........................................................................x FOREWORD ................................................................................................................................. xi AUTHORS, CONTRIBUTORS, AND REVIEWERS ................................................................ xii -

Synthesis of Organobromines As a Tool for Their Characterisation and Environmental Occurrence Assessment
Synthesis of organobromines as a tool for their characterisation and environmental occurrence assessment Andreas Rydén Department of Materials and Environmental Chemistry Stockholm University Stockholm 2013 i Doctoral Thesis 2013 Department of Materials and Environmental Chemistry Stockholm University SE-106 91 Stockholm Sweden Abstract Polybrominated diphenyl ethers (PBDEs) have been intensively used as flame retardants (FRs) and have become ubiquitous environmental pollutants. PBDEs form hydroxylated PBDEs (OH-PBDEs) as metabolites. Further, some OH-PBDEs and methoxy-PBDEs (MeO-PBDEs) are natural products. These are all compounds of environmental and health concern and it is therefore important to confirm their identity and to assess their environmental levels and toxicities. Hence, it is vital to obtain authentic reference standards of individual PBDEs and OH/MeO-PBDEs. The thesis main aim was to develop synthesis methods of congener specific PBDEs, OH- and MeO-PBDEs. The second aim was to identify and quantify PBDEs, OH- and MeO-PBDEs in environmental samples. The third was to propose an abbreviation system for FRs. O-Arylation of brominated phenols, using either symmetrical or unsymmetrical brominated diphenyliodonium salts, was selected for synthesis of PBDEs and OH- /MeO-PBDEs. A total of 16 MeO-PBDEs, 11 OH-PBDEs, 1 diMeO-PBDE and 1 EtO-MeO-PBDE were synthesised. Three novel unsymmetrical diaryliodonium triflates were synthesised and used in synthesis. Optimisations were made to construct a reliable general method for congener specific PBDE synthesis, which was used in the synthesis of 8 representative PBDE congeners. The products were generally characterised by electron ionisation mass spectrometry (EIMS) and nuclear magnetic resonance (NMR) spectroscopy. -

LABORATORY 9 the Grignard Reaction: a Microscale Preparation
Organic Chemistry: Techniques and Transformations LABORATORY 9 The Grignard Reaction: A Microscale Preparation of Benzoic Acid Introduction Your laboratory skills have grown considerably since the first of the semester, and you are ready for the challenge of a famous reaction--one marked by unusual materials and striking chemical and physical changes. Try, especially, to understand exactly which chemical structure and equation corresponds to each stage of this dramatic synthesis. Your work in this laboratory is a departure from your previous experience. You will work on a micro scale. Microscale apparatus is used throughout the second semester organic laboratories (CHEM 2130). Working on a microscale greatly reduces both the quantities of material and the reaction time. The regular scale reaction requires nearly two full periods. The microscale work can be done in one period. Further, the reduced scale minimizes the hazard of working with ether, a very flammable solvent with a low flash point. You must eliminate flames and sparks when working with ether. To run a successful Grignard reaction, it is essential that moisture (including water vapor from the air) be excluded from the reaction apparatus. Septa (rubber caps) on the reagent bottles and on the reaction tubes effectively exclude moisture, so that a calcium sulfate drying tube is unnecessary. A syringe, with a needle, is used to remove reagents from septum protected storage bottles and to add the reagent to a reaction tube. Thus, use of a syringe and septum eliminates the need for an addition funnel and a costly three-necked round-bottomed flask. There is less set-up time, less clean-up time and less breakage of expensive glassware. -

Synthesis of Novel Azetidines
University of New Orleans ScholarWorks@UNO University of New Orleans Theses and Dissertations Dissertations and Theses Fall 12-20-2013 Synthesis of Novel Azetidines Amber Thaxton UNO, [email protected] Follow this and additional works at: https://scholarworks.uno.edu/td Part of the Medicinal and Pharmaceutical Chemistry Commons, and the Organic Chemistry Commons Recommended Citation Thaxton, Amber, "Synthesis of Novel Azetidines" (2013). University of New Orleans Theses and Dissertations. 1764. https://scholarworks.uno.edu/td/1764 This Dissertation is protected by copyright and/or related rights. It has been brought to you by ScholarWorks@UNO with permission from the rights-holder(s). You are free to use this Dissertation in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you need to obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Dissertation has been accepted for inclusion in University of New Orleans Theses and Dissertations by an authorized administrator of ScholarWorks@UNO. For more information, please contact [email protected]. Synthesis of Novel Azetidines A Dissertation Submitted to the Graduate Faculty of the University of New Orleans in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Chemistry by Amber Nichole Thaxton B.S. Clemson University, 2004 M.S. University of New Orleans, 2013 December 2013 For my family: To my mom, Bee Thaxton, for everything she has done for me. I am extremely thankful for her constant support and reassurance. -

Safety Data Sheets
SAFETY DATA SHEETS According to Globally Harmonized System of Classification and Labelling of Chemicals (GHS) - Sixth revised edition Version: 1.0 Creation Date: Feb. 6, 2018 Revision Date: Feb. 6, 2018 1. Identification 1.1 GHS Product identifier Product name Bromobenzene 1.2 Other means of identification Product number A502171 Other names phenyl benzyl thioether 1.3 Recommended use of the chemical and restrictions on use Identified uses Used for research and development only. Uses advised against no data available 1.4 Supplier's details Company Sangon Biotech (Shanghai) Co., Ltd. Address 698 Xiangmin Road, Songjiang, Shanghai 201611, China Telephone +86-400-821-0268 / +86-800-820-1016 Fax +86-400-821-0268 to 9 1.5 Emergency phone number Emergency phone +86-21-57072055 number Service hours Monday to Friday, 9am-5pm (Standard time zone: UTC/GMT +8 hours). 2. Hazard identification 2.1 Classification of the substance or mixture Flammable liquids, Category 3 Skin irritation, Category 2 Hazardous to the aquatic environment, long-term (Chronic) - Category Chronic 2 2.2 GHS label elements, including precautionary statements Pictogram(s) Bromobenzene Page 1 of 9 Signal word Warning Hazard statement(s) H226 Flammable liquid and vapour H315 Causes skin irritation H411 Toxic to aquatic life with long lasting effects Precautionary statement(s) Prevention P210 Keep away from heat, hot surfaces, sparks, open flames and other ignition sources. No smoking. P233 Keep container tightly closed. P240 Ground and bond container and receiving equipment. P241 Use explosion-proof [electrical/ventilating/lighting/...] equipment. P242 Use non-sparking tools. P243 Take action to prevent static discharges. -
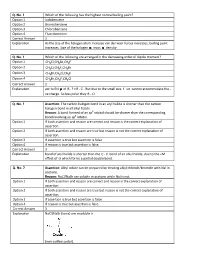
Q. No. 3 Which of the Following Has the Highest Normal Boiling Point
Q. No. 3 Which of the following has the highest normal boiling point? Option 1 Iodobenzene Option 2 Bromobenzene Option 3 Chlorobenzene Option 4 Fluorobenzene Correct Answer 1 Explanation As the size of the halogen atom increase van der waal forces increases, boiling point increases. Size of the halogen mass density. Q. No. 4 Which of the following are arranged in the decreasing order of dipole moment? Option 1 CH3 Cl,CH 3 Br,CH 3 F Option 2 CH3 Cl,CH 3 F,CH 3 Br Option 3 CH3 Br,CH 3 Cl,CH 3 F Option 4 CH3 Br,CH 3 F,CH 3 Cl Correct Answer 2 Explanation acc to EN of RF> RCl . But due to the small size. F ion cannot accommodate the ve charge. So less polar they R Cl . Q. No. 5 Assertion : The carbon halogen bond in an aryl halid e is shorter than the carbon halogen bond in all alkyl halide. Reason : A bond formed of an sp 3 orbital should be shorter than the corresponding bond involving an sp 2 orbital. Option 1 If both assertion and reason are correct and reason is the correct explanation of assertion. Option 2 If both assertion and reason are true but reason is not the correct explanation of assertion. Option 3 If assertion is true but assertion is false. Option 4 If reason is true but assertion is false. Correct Answer 3 Explanation bond of aryl halide is shorter than the C X bond of an alkyl halide, due to the +M effect of X which forms a partial double bond. -

Synthesis and Evaluation of Novel Tropane Compounds As Potential Therapeutics for Drug Abuse
University of New Orleans ScholarWorks@UNO University of New Orleans Theses and Dissertations Dissertations and Theses 8-8-2007 Synthesis And Evaluation Of Novel Tropane Compounds As Potential Therapeutics For Drug Abuse Harneet Kaur University of New Orleans Follow this and additional works at: https://scholarworks.uno.edu/td Recommended Citation Kaur, Harneet, "Synthesis And Evaluation Of Novel Tropane Compounds As Potential Therapeutics For Drug Abuse" (2007). University of New Orleans Theses and Dissertations. 586. https://scholarworks.uno.edu/td/586 This Dissertation is protected by copyright and/or related rights. It has been brought to you by ScholarWorks@UNO with permission from the rights-holder(s). You are free to use this Dissertation in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you need to obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Dissertation has been accepted for inclusion in University of New Orleans Theses and Dissertations by an authorized administrator of ScholarWorks@UNO. For more information, please contact [email protected]. Synthesis And Evaluation Of Novel Tropane Compounds As Potential Therapeutics For Drug Abuse A Dissertation Submitted to the Graduate Faculty of the University of New Orleans in partial fulfillment of the requirements for the degree of Doctor of Philosophy in The Department of Chemistry by Harneet Kaur M.S. (Hons) Panjab University 2001 M.S. University of New Orleans 2004 August 2007 To my family, for their love and support Father: Dr. -
LIQUID VISCOSITY and CHEMICAL CONSTITUTION of ORGANIC COMPOUNDS : a NEW CORRELATION Mmw and a COMPILATION of LITERATURE DATA
vìi ιΤ»Αϋ.ΓΪ*>*ν >tíÀí.k *ΊιπίιΠ IFIULJ-V.!·» 'ft' Λ'. -låiiSiip ¡Ili^iilfeÉ COMMISSION OF THE EUROPEAN COMMUNITIES ■Mié LIQUID VISCOSITY AND CHEMICAL CONSTITUTION OF ORGANIC COMPOUNDS : A NEW CORRELATION mMW AND A COMPILATION OF LITERATURE DATA by D. VAN VELZEN (Euratom) R. LOPES CARDOZO (A.K.Z.O., Albatros Koninklijke Zout Organon, Hengelo, the Netherlands) H. LANGENKAMP (Euratom) 1972 Joint Nuclear Research Centre Ispra Establishment Italy i Chemistry wmSËSS^gSSXÊÊ. m -li LEGAL NOTICE iiíliip mm•m' This document was prepared under the sponsorship of the CommissioM'Ai n of the European Communities. mr 'MÈwffi!mw-'< Neither the Commission of the European Communities, its contractors nor any person acting on their behalf: make any warranty or representation, express or implied, with respect to the accuracy, completeness, or usefulness of the information contained in this document, or that the use of any information, apparatus, method or process disclosed in this document may not infringe privately owned rights; or assume any liability.mtmmEEm with respect to the use of, or fo r damages resultin11.11g $ from the use of any information, apparatus, method or process disclosed in this document. «If ;-«Thi s report is on sale at the addresses listed on cover page iliPili4 i ¡Éill¡p^ ommission of the Europeanui upean Communitievjommunmes D.G. XIII C.I.D. {VTI-f Ïm4w 29, rue Aldringen xembourg Jw i $m M ,**■ ii '.»■>« 'f f F* ■*[..-■: UI: J Uni f||i »sira This document was reproduced on the basis of the best available copy. mm ιΛ« EUR 4735 e LIQUID VISCOSITY AND CHEMICAL CONSTITUTION OF ORGANIC COMPOUNDS : A NEW CORRELATION AND A COMPILATION OF LITERATURE DATA by D.