General Pathology Lectures
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Job Posting Clinical Microbiology Final
The Department of Pathology & Cell Biology at Columbia University Irving Medical Center (CUIMC) is recruiting for an MD, MD/PhD, or PhD academic clinical microbiologist of any rank to join our faculty as a Medical Director of the NewYork-Presbyterian/CUIMC Clinical Microbiology Laboratory. Applicants should have an established track record of accomplishment within the field of clinical microbiology and a demonstrated ability to lead an experienced group of laboratory technologists, supervisors, and staff. In addition to strong clinical and technical skills, particular emphasis is placed on candidates with a demonstrated record of collegiality and inter-departmental collaboration. Applicants must have completed a fellowship in clinical microbiology and be board-certified/board-eligible in Medical and Public Health Microbiology through the American Board of Medical Microbiology (ABMM) or board-certified/board- eligible in Clinical Pathology with subspecialty certification in Medical Microbiology through the American Board of Pathology (ABP). The applicant must also be able to satify clinical licensing requirements to serve as a Laboratory Director in New York State. The successful applicant will help oversee diagnostic testing in the areas of Bacteriology, Virology, Mycobacteriology, Mycology, and Parasitology. The position also includes responsibilities for teaching of pathology residents, medical students, infectious diseases fellows, and technical staff. Applicants must be currently involved in ongoing research with a track record of publications in the field. The position offers a competitive salary commensurate with training and experience, and an appointment to the faculty of the Columbia University Vagelos College of Physicians & Surgeons. The Clinical Microbiology Laboratory at NewYork-Presbyterian/CUIMC is located in the Washington Heights neighborhood of New York City, offering unparalleled opportunities to work and live in a thriving, diverse, metropolitan environment with access to world-class cultural institutions, restaurants, and entertainment. -

Chlamydia Trachomatis Infection Is Driven by Nonprotective Immune Cells That Are Distinct from Protective Populations
Pathology after Chlamydia trachomatis infection is driven by nonprotective immune cells that are distinct from protective populations Rebeccah S. Lijeka,b,1, Jennifer D. Helblea, Andrew J. Olivea,c, Kyra W. Seigerb, and Michael N. Starnbacha,1 aDepartment of Microbiology and Immunobiology, Harvard Medical School, Boston, MA 02115; bDepartment of Biological Sciences, Mount Holyoke College, South Hadley, MA 01075; and cDepartment of Microbiology and Physiological Systems, University of Massachusetts Medical School, Worcester, MA 01605 Edited by Rafi Ahmed, Emory University, Atlanta, GA, and approved December 27, 2017 (received for review June 23, 2017) Infection with Chlamydia trachomatis drives severe mucosal immu- sequence identity, Chlamydia muridarum, the extent to which the nopathology; however, the immune responses that are required for molecular pathogenesis of C. muridarum represents that of Ct is mediating pathology vs. protection are not well understood. Here, unknown (6). Ct serovar L2 (Ct L2) is capable of infecting the we employed a mouse model to identify immune responses re- mouse upper genital tract when inoculated across the cervix into quired for C. trachomatis-induced upper genital tract pathology the uterus (7, 8) but it does not induce robust immunopathology. and to determine whether these responses are also required for This is consistent with the human disease phenotype caused by Ct L2, bacterial clearance. In mice as in humans, immunopathology was which disseminates to the lymph nodes causing lymphogranuloma characterized by extravasation of leukocytes into the upper genital venereum (LGV) and is not a major cause of mucosal immunopa- thology in the female upper genital tract (uterus and ovaries). tract that occluded luminal spaces in the uterus and ovaries. -

Printable Version
Clinical Pathology Laboratories Drugs of Abuse Testing Offering our clients state-of-the-art testing is part of CPL’s ongoing commitment to excellence. Effective 06/18/2018, Clinical Pathology Laboratories (CPL) will replace current drug of abuse (DAU) screening and screening with reflex confirmation profiles. The new profiles provide: • Reformulation and recombination of classes for profile testing, more relevant to current societal trends • Standardization of drug thresholds to high sensitivity cutoff values • Addition of the following analytes in specific profiles for testing: o Fentanyl o Buprenorphine o 6-Acetylmorphine o MDMA (Ecstasy) • Expansion of testing for adulterants with disqualifying comments added to specimens determined to be altered • Expansion of interpretive notes on reports regarding the method, cutoff values, and specific drugs that may or may not be detected by the screening method New Profile Name EtOH Opiates Cocaine Fentanyl Oxycodone Order Code Methadone Barbiturates Cannabinoids Phencyclidine Acetylmorphine Buprenorphine Amphetamines MDMA/Ecstasy - Benzodiazepines 6 Drug of Abuse, 8 Analytes 3305 X X X X X X X X No Confirm Drug of Abuse, 8 Analytes 3306 X X X X X X X X w/ Confirm Drug of Abuse, 9 Analytes 3316 X X X X X X X X X w/ EtOH, No Confirm Drug of Abuse, 9 Analytes 3315 X X X X X X X X X w/ EtOH, w/ Confirm Drug of Abuse, 9 Analytes 3307 X X X X X X X X X No THC or Confirm Drug of Abuse, 9 Analytes 3308 X X X X X X X X X No THC, w/ Confirm Drug of Abuse, 10 Analytes 3317 X X X X X X X X X X No Confirm -

Clinical Pathology, Immunopathology and Advanced Vaccine Technology in Bovine Theileriosis: a Review
pathogens Review Clinical Pathology, Immunopathology and Advanced Vaccine Technology in Bovine Theileriosis: A Review Onyinyechukwu Ada Agina 1,2,* , Mohd Rosly Shaari 3, Nur Mahiza Md Isa 1, Mokrish Ajat 4, Mohd Zamri-Saad 5 and Hazilawati Hamzah 1,* 1 Department of Veterinary Pathology and Microbiology, Faculty of Veterinary Medicine, Universiti Putra Malaysia, Serdang 43400, Malaysia; [email protected] 2 Department of Veterinary Pathology and Microbiology, Faculty of Veterinary Medicine, University of Nigeria Nsukka, Nsukka 410001, Nigeria 3 Animal Science Research Centre, Malaysian Agricultural Research and Development Institute, Headquarters, Serdang 43400, Malaysia; [email protected] 4 Department of Veterinary Pre-clinical sciences, Faculty of Veterinary Medicine, Universiti Putra Malaysia, Serdang 43400, Malaysia; [email protected] 5 Research Centre for Ruminant Diseases, Faculty of Veterinary Medicine, Universiti Putra Malaysia, Serdang 43400, Malaysia; [email protected] * Correspondence: [email protected] (O.A.A.); [email protected] (H.H.); Tel.: +60-11-352-01215 (O.A.A.); +60-19-284-6897 (H.H.) Received: 2 May 2020; Accepted: 16 July 2020; Published: 25 August 2020 Abstract: Theileriosis is a blood piroplasmic disease that adversely affects the livestock industry, especially in tropical and sub-tropical countries. It is caused by haemoprotozoan of the Theileria genus, transmitted by hard ticks and which possesses a complex life cycle. The clinical course of the disease ranges from benign to lethal, but subclinical infections can occur depending on the infecting Theileria species. The main clinical and clinicopathological manifestations of acute disease include fever, lymphadenopathy, anorexia and severe loss of condition, conjunctivitis, and pale mucous membranes that are associated with Theileria-induced immune-mediated haemolytic anaemia and/or non-regenerative anaemia. -

Department of Experimental Pathology, Immunology and Microbiology 531
Department of Experimental Pathology, Immunology and Microbiology 531 Department of Experimental Pathology, Immunology and Microbiology Interim Chairperson: Zaatari, Ghazi Vice Chairperson: Matar, Ghassan Professors: Abdelnoor, Alexander; Khouri, Samia; Matar, Ghassan; Sayegh, Mohamed; Zaatari, Ghazi Associate Professor: Rahal, Elias Assistant Professors: Al-Awar, Ghassan; El Hajj, Hiba; Shirinian, Margret; Zaraket, Hassan The Department of Experimental Pathology, Immunology and Microbiology offers courses to medical laboratory sciences (MLSP) students as well as nursing, medical, and graduate students. It offers a graduate program (discipline of Microbiology and Immunology) leading to a master’s degree (MS) or doctoral degree (PhD) in Biomedical sciences. The requirements for admission to the graduate program are stated on page 33 of this catalogue. IDTH 203 The immune System in Health and Disease 37.28; 3 cr. See Interdepartmental Courses. IDTH 205 Microbiology and Infectious Diseases 37.28; 5 cr. See Interdepartmental Courses. MBIM 223 Parasitology for MLSP Students 39.39; 4 cr. Second semester. MBIM 237 Microbiology and Immunology for Nursing Degree Students 32.64; 3 cr. A course on the fundamental aspects of medical microbiology and immunology for nursing students. Second semester. MBIM 260 Elective in Infectious Diseases for Medicine III and IV 0.180 A course on basic evaluation, diagnosis, and management of infectious diseases. One month. MBIM 261 Elective in Immunology for Medicine III and IV 0.180 A course that is an introduction to immunological research and its application to clinical practice. One month. MBIM 310 Basic Immunology 32.32; 3 cr. A course on innate and adaptive immune mechanisms, infection and immunity, vaccination, immune mechanisms in tissue injury and therapeutic immunology. -

Deciphering the Triad of Infection, Immunity and Pathology
INSIGHT DISEASE Deciphering the triad of infection, immunity and pathology The factors which drive and control disease progression can be inferred from mathematical models that integrate measures of immune responses, data from tissue sampling and markers of infection dynamics. FREDERIK GRAW immune actors in the body. Now, in eLife, Related research article Myers MA, Smith Amber Smith and colleagues at St. Jude Child- AP, Lane LC, Moquin DJ, Aogo R, Woolard ren’s Research Hospital, the University of Ten- S, Thomas P, Vogel P, Smith AM. 2021. nessee Health Science Center and the Dynamically linking influenza virus infection Washington University School of Medicine – kinetics, lung injury, inflammation, and dis- including Margaret Myers and Amanda Smith as ease severity. eLife 10:e68864. doi: 10. joing first authors – report how viral infection, 7554/eLife.68864 counteracting immune responses and lung pathology interact as mice fight off influenza A (Myers et al., 2021). First, the team tracked how viral load and the number of CD8+ T cells, an important immune fever, a cough, a splitting headache... actor that helps to clear infected cells, pro- Being sick often comes with tell-tale gressed over time. In combination with mathe- A signs which worsen as the disease pro- matical models, these measurements allowed gresses and tissues become damaged. These Myers et al. to estimate several parameters that symptoms result from complex interactions reflect the pace at which the virus replicates, the between the infecting pathogen, the inflamma- strength of the immune response, and the inter- tion process, and the response from the immune actions between these processes. -

Overview of Pathology and Its Related Disciplines - Soheir Mahmoud Mahfouz
MEDICAL SCIENCES – Vol.I -Overview of Pathology and its Related Disciplines - Soheir Mahmoud Mahfouz OVERVIEW OF PATHOLOGY AND ITS RELATED DISCIPLINES Soheir Mahmoud Mahfouz Cairo University, Kasr El Ainy Hospital, Egypt Keywords: Pathology, Pathology disciplines, Pathology techniques, Ancillary diagnostic methods, General Pathology, Special Pathology Contents 1. Introduction 1.1 Pathology coverage 1.1.1 Etiology and Pathogenesis of a Disease 1.1.2 Manifestations of Disease (Lesions) 1.1.3 Phases Of A Disease Process (Course) 1.2 Physician’s approach to patient 1.3 Types of pathologists and affiliated specialties 1.4 Role of pathologist 2. Pathology and its related disciplines 2.1 Cytology 2.1.1 Cytology Samples 2.1.2 Technical Aspects 2.1.3 Examination of Sample and Diagnosis 3. Pathology techniques and ancillary diagnostic methods 3.1 Macroscopic pathology 3.2 Light Microscopy 3.3 Polarizing light microscopy 3.4 Electron microscopy (EM) 3.5 Confocal Microscopy 3.6 Frozen section 3.7 Cyto/histochemistry 3.8 Immunocyto/histochemical methods 3.9 Molecular and genetic methods of diagnosis 3.10 Quantitative methods 4. Types of tests used in pathology 4.1 DiagnosticUNESCO tests – EOLSS 4.2 Quantitative tests 4.3 Prognostic tests 5. The scope of SAMPLEpathology & its main divisions CHAPTERS 6. Conclusions Glossary Bibliography Biographical sketch Summary Pathology is the science of disease. It deals with deviations from normal body function and ©Encyclopedia of Life Support Systems (EOLSS) MEDICAL SCIENCES – Vol.I -Overview of Pathology and its Related Disciplines - Soheir Mahmoud Mahfouz structure. Many disciplines are involved in the study of disease, as it is necessary to understand the complex causes and effects of various disorders that affect the organs and body as a whole. -

American Society for Clinical Pathology to the Clinical Laboratory Improvement Advisory Committee
Statement from the American Society for Clinical Pathology to the Clinical Laboratory Improvement Advisory Committee The American Society for Clinical Pathology is pleased to provide this statement to the Clinical Laboratory Improvement Advisory Committee (CLIAC) on the roles, responsibilities and competencies of bioinformaticists. The completion of the Human Genome Project has resulted in vast sums of patient data, and bioinformaticists are increasingly being utilized by clinical laboratories to manage, process, and analyze it, especially in the rapidly expanding specialty of molecular diagnostics. Bioinformaticists, and the unique skills these individuals bring, are also helping to transform the practice of pathology and laboratory medicine by developing or/or enhancing the bioinformatics tools used to expand the ability of pathology and laboratory medicine to protect patient health. ASCP greatly appreciates CLIAC’s leadership by focusing attention on the valuable contribution these professionals are making and to improve their ability to do so. The following comments are based on comments provided by our membership during our efforts to respond to the questions posed by the CLIAC. CLIAC Discussion Questions: Question 1: Are Bioinformaticists needed in clinical and public health laboratories? If so, what are the current roles, responsibilities, and competencies of bioinformaticists in these settings? ASCP believes that bioinformaticists are a key component of high quality, full service clinical laboratories, though the roles and responsibilities of these professionals may vary significantly. Informaticists are critical to building the bioinformatics pipeline, which can include the software and database engineering, configuration of available bioinformatics software, and/or management and interfacing of LIS and other informatics systems, both internally within the laboratory (e.g. -

Neuropathic Pain: Delving Into the Oxidative Origin and the Possible Implication of Transient Receptor Potential Channels
REVIEW published: 14 February 2018 doi: 10.3389/fphys.2018.00095 Neuropathic Pain: Delving into the Oxidative Origin and the Possible Implication of Transient Receptor Potential Channels Cristina Carrasco 1*, Mustafa Nazirogluˇ 2, Ana B. Rodríguez 1 and José A. Pariente 1 1 Department of Physiology, Faculty of Sciences, University of Extremadura, Badajoz, Spain, 2 Neuroscience Research Center, Suleyman Demirel University, Isparta, Turkey Currently, neuropathic pain is an underestimated socioeconomic health problem affecting millions of people worldwide, which incidence may increase in the next years due to chronification of several diseases, such as cancer and diabetes. Growing evidence links neuropathic pain present in several disorders [i.e., spinal cord injury (SCI), cancer, diabetes and alcoholism] to central sensitization, as a global result of mitochondrial dysfunction induced by oxidative and nitrosative stress. Additionally, inflammatory signals Edited by: and the overload in intracellular calcium ion could be also implicated in this complex Ali Mobasheri, network that has not yet been elucidated. Recently, calcium channels namely transient University of Surrey, United Kingdom receptor potential (TRP) superfamily, including members of the subfamilies A (TRAP1), M Reviewed by: Felipe Simon, (TRPM2 and 7), and V (TRPV1 and 4), have demonstrated to play a role in the nociception Universidad Andrés Bello, Chile mediated by sensory neurons. Therefore, as neuropathic pain could be a consequence of Enrique Soto, the imbalance between reactive oxygen species and endogen antioxidants, antioxidant Benemérita Universidad Autónoma de Puebla, Mexico supplementation may be a treatment option. This kind of therapy would exert its beneficial *Correspondence: action through antioxidant and immunoregulatory functions, optimizing mitochondrial Cristina Carrasco function and even increasing the biogenesis of this vital organelle; on balance, antioxidant [email protected] supplementation would improve the patient’s quality of life. -
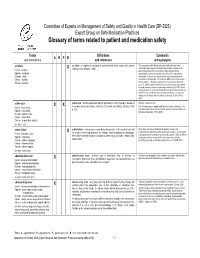
Glossary of Terms Related to Patient and Medication Safety
Committee of Experts on Management of Safety and Quality in Health Care (SP-SQS) Expert Group on Safe Medication Practices Glossary of terms related to patient and medication safety Terms Definitions Comments A R P B and translations and references and synonyms accident accident : an unplanned, unexpected, and undesired event, usually with adverse “For many years safety officials and public health authorities have Xconsequences (Senders, 1994). discouraged use of the word "accident" when it refers to injuries or the French : accident events that produce them. An accident is often understood to be Spanish : accidente unpredictable -a chance occurrence or an "act of God"- and therefore German : Unfall unavoidable. However, most injuries and their precipitating events are Italiano : incidente predictable and preventable. That is why the BMJ has decided to ban the Slovene : nesreča word accident. (…) Purging a common term from our lexicon will not be easy. "Accident" remains entrenched in lay and medical discourse and will no doubt continue to appear in manuscripts submitted to the BMJ. We are asking our editors to be vigilant in detecting and rejecting inappropriate use of the "A" word, and we trust that our readers will keep us on our toes by alerting us to instances when "accidents" slip through.” (Davis & Pless, 2001) active error X X active error : an error associated with the performance of the ‘front-line’ operator of Synonym : sharp-end error French : erreur active a complex system and whose effects are felt almost immediately. (Reason, 1990, This definition has been slightly modified by the Institute of Medicine : “an p.173) error that occurs at the level of the frontline operator and whose effects are Spanish : error activo felt almost immediately.” (Kohn, 2000) German : aktiver Fehler Italiano : errore attivo Slovene : neposredna napaka see also : error active failure active failures : actions or processes during the provision of direct patient care that Since failure is a term not defined in the glossary, its use is not X recommended. -

2021 Anatomic & Clinical Pathology
BEAUMONT LABORATORY 2021 ANATOMIC & CLINICAL PATHOLOGY Physician Biographies Expertise BEAUMONT LABORATORY • 800-551-0488 BEAUMONT LABORATORY ANATOMIC & CLINICAL PATHOLOGY • PHYSICIAN BIOGRAPHIES Peter Millward, M.D. Mitual Amin, M.D. Chief of Clinical Pathology, Beaumont Health Interim Chair, Pathology and Laboratory Medicine, Interim Chief of Pathology Service Line, Beaumont Health Royal Oak Interim Physician Executive, Beaumont Medical Group Interim Chair, Department of Pathology and Laboratory Medicine, Oakland University William Beaumont School Interim System Medical Director, Beaumont Laboratory of Medicine Outreach Services Board certification Associate Medical Director, Blood Bank and • Anatomic and Clinical Pathology, Transfusion Medicine, Beaumont Health American Board of Pathology Board certification Additional fellowship training • Anatomic and Clinical Pathology, • Surgical Pathology American Board of Pathology Special interests Subspecialty board certification • Breast Pathology, Genitourinary Pathology, • Blood Banking and Transfusion Medicine, Gastrointestinal Pathology American Board of Pathology Lubna Alattia, M.D. Kurt D. Bernacki, M.D. Cytopathologist and Surgical Pathologist, Trenton System Medical Director, Surgical Pathology Board certification Beaumont Health • Anatomic and Clinical Pathology, Chief, Pathology Laboratory, West Bloomfield American Board of Pathology Breast Care Center Subspecialty board certification Diagnostic Lead, Pulmonary Tumor Pathology • Cytopathology, American Board of Pathology Diagnostic -
Pathology: a Career in Medicine the Study of the Nature of Disease, Its Causes, Processes, Development, and Consequences
PATHOLOGY A Career in Medicine The Intersociety Council for Pathology Information (ICPI) www.pathologytraining.org 2015 Pathology: A Career in Medicine The study of the nature of disease, its causes, processes, development, and consequences. Pathology is the medical specialty that provides a scientific foundation for medical practice The pathologist is a physician who specializes in the diagnosis and management of human disease by laboratory methods. Pathologists function in three broad areas: as diagnosticians, as teachers, and as investigators. Fundamental to the discipline of pathology is the need to integrate clinical information with physiological, biochemical and molecular laboratory studies, together with observations of tissue alterations. Pathologists in hospital and clinical laboratories practice as consultant physicians, developing and applying knowledge of tissue and laboratory analyses to assist in the diagnosis and treatment of individual patients. As teachers, they impart this knowledge of disease to their medical colleagues, to medical students, and to trainees at all levels. As scientists, they use the tools of laboratory science in clinical studies, disease models, and other experimental systems, to advance the understanding and treatment of disease. Pathology has a special appeal to those who enjoy solving disease-related problems, using technologies based upon fundamental sciences ranging from biophysics to molecular genetics, as well as tools from the more traditional disciplines of anatomy, biochemistry, pharmacology, physiology and microbiology. The Pathologist in Patient Care The pathologist uses diagnostic and screening tests to identify and interpret the changes that characterize different diseases in the cells, tissues, and fluids of the body. Anatomic pathology involves the analysis of the A biosample robot prepares specimens for gross and microscopic structural changes caused by testing disease in tissues and cells removed during biopsy procedures, in surgery, or at autopsy.