Structural Basis of Inhibition of the Pioneer Transcription Factor NF-Y by Suramin
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Gene Expression Data for Gene Ontology
ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION A Thesis Presented to The Graduate Faculty of The University of Akron In Partial Fulfillment of the Requirements for the Degree Master of Science Robert Daniel Macholan May 2011 ANALYSIS OF GENE EXPRESSION DATA FOR GENE ONTOLOGY BASED PROTEIN FUNCTION PREDICTION Robert Daniel Macholan Thesis Approved: Accepted: _______________________________ _______________________________ Advisor Department Chair Dr. Zhong-Hui Duan Dr. Chien-Chung Chan _______________________________ _______________________________ Committee Member Dean of the College Dr. Chien-Chung Chan Dr. Chand K. Midha _______________________________ _______________________________ Committee Member Dean of the Graduate School Dr. Yingcai Xiao Dr. George R. Newkome _______________________________ Date ii ABSTRACT A tremendous increase in genomic data has encouraged biologists to turn to bioinformatics in order to assist in its interpretation and processing. One of the present challenges that need to be overcome in order to understand this data more completely is the development of a reliable method to accurately predict the function of a protein from its genomic information. This study focuses on developing an effective algorithm for protein function prediction. The algorithm is based on proteins that have similar expression patterns. The similarity of the expression data is determined using a novel measure, the slope matrix. The slope matrix introduces a normalized method for the comparison of expression levels throughout a proteome. The algorithm is tested using real microarray gene expression data. Their functions are characterized using gene ontology annotations. The results of the case study indicate the protein function prediction algorithm developed is comparable to the prediction algorithms that are based on the annotations of homologous proteins. -

1 Supporting Information for a Microrna Network Regulates
Supporting Information for A microRNA Network Regulates Expression and Biosynthesis of CFTR and CFTR-ΔF508 Shyam Ramachandrana,b, Philip H. Karpc, Peng Jiangc, Lynda S. Ostedgaardc, Amy E. Walza, John T. Fishere, Shaf Keshavjeeh, Kim A. Lennoxi, Ashley M. Jacobii, Scott D. Rosei, Mark A. Behlkei, Michael J. Welshb,c,d,g, Yi Xingb,c,f, Paul B. McCray Jr.a,b,c Author Affiliations: Department of Pediatricsa, Interdisciplinary Program in Geneticsb, Departments of Internal Medicinec, Molecular Physiology and Biophysicsd, Anatomy and Cell Biologye, Biomedical Engineeringf, Howard Hughes Medical Instituteg, Carver College of Medicine, University of Iowa, Iowa City, IA-52242 Division of Thoracic Surgeryh, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada-M5G 2C4 Integrated DNA Technologiesi, Coralville, IA-52241 To whom correspondence should be addressed: Email: [email protected] (M.J.W.); yi- [email protected] (Y.X.); Email: [email protected] (P.B.M.) This PDF file includes: Materials and Methods References Fig. S1. miR-138 regulates SIN3A in a dose-dependent and site-specific manner. Fig. S2. miR-138 regulates endogenous SIN3A protein expression. Fig. S3. miR-138 regulates endogenous CFTR protein expression in Calu-3 cells. Fig. S4. miR-138 regulates endogenous CFTR protein expression in primary human airway epithelia. Fig. S5. miR-138 regulates CFTR expression in HeLa cells. Fig. S6. miR-138 regulates CFTR expression in HEK293T cells. Fig. S7. HeLa cells exhibit CFTR channel activity. Fig. S8. miR-138 improves CFTR processing. Fig. S9. miR-138 improves CFTR-ΔF508 processing. Fig. S10. SIN3A inhibition yields partial rescue of Cl- transport in CF epithelia. -

Molecular Mechanisms of Ribosomal Protein Gene Coregulation
Downloaded from genesdev.cshlp.org on October 3, 2021 - Published by Cold Spring Harbor Laboratory Press Molecular mechanisms of ribosomal protein gene coregulation Rohit Reja, Vinesh Vinayachandran, Sujana Ghosh, and B. Franklin Pugh Center for Eukaryotic Gene Regulation, Pennsylvania State University, University Park, Pennsylvania 16802, USA The 137 ribosomal protein genes (RPGs) of Saccharomyces provide a model for gene coregulation. We examined the positional and functional organization of their regulators (Rap1 [repressor activator protein 1], Fhl1, Ifh1, Sfp1, and Hmo1), the transcription machinery (TFIIB, TFIID, and RNA polymerase II), and chromatin at near-base-pair res- olution using ChIP-exo, as RPGs are coordinately reprogrammed. Where Hmo1 is enriched, Fhl1, Ifh1, Sfp1, and Hmo1 cross-linked broadly to promoter DNA in an RPG-specific manner and demarcated by general minor groove widening. Importantly, Hmo1 extended 20–50 base pairs (bp) downstream from Fhl1. Upon RPG repression, Fhl1 remained in place. Hmo1 dissociated, which was coupled to an upstream shift of the +1 nucleosome, as reflected by the Hmo1 extension and core promoter region. Fhl1 and Hmo1 may create two regulatable and positionally distinct barriers, against which chromatin remodelers position the +1 nucleosome into either an activating or a repressive state. Consistent with in vitro studies, we found that specific TFIID subunits, in addition to cross-linking at the core promoter, made precise cross-links at Rap1 sites, which we interpret to reflect native Rap1–TFIID interactions. Our findings suggest how sequence-specific DNA binding regulates nucleosome positioning and transcription complex assembly >300 bp away and how coregulation coevolved with coding sequences. -

Novel Candidate Genes of Thyroid Tumourigenesis Identified in Trk-T1 Transgenic Mice
Endocrine-Related Cancer (2012) 19 409–421 Novel candidate genes of thyroid tumourigenesis identified in Trk-T1 transgenic mice Katrin-Janine Heiliger*, Julia Hess*, Donata Vitagliano1, Paolo Salerno1, Herbert Braselmann, Giuliana Salvatore 2, Clara Ugolini 3, Isolde Summerer 4, Tatjana Bogdanova5, Kristian Unger 6, Gerry Thomas6, Massimo Santoro1 and Horst Zitzelsberger Research Unit of Radiation Cytogenetics, Helmholtz Zentrum Mu¨nchen, Ingolsta¨dter Landstr. 1, 85764 Neuherberg, Germany 1Istituto di Endocrinologia ed Oncologia Sperimentale del CNR, c/o Dipartimento di Biologia e Patologia Cellulare e Molecolare, Universita` Federico II, Naples 80131, Italy 2Dipartimento di Studi delle Istituzioni e dei Sistemi Territoriali, Universita` ‘Parthenope’, Naples 80133, Italy 3Division of Pathology, Department of Surgery, University of Pisa, 56100 Pisa, Italy 4Institute of Radiation Biology, Helmholtz Zentrum Mu¨nchen, 85764 Neuherberg, Germany 5Institute of Endocrinology and Metabolism, Academy of Medical Sciences of the Ukraine, 254114 Kiev, Ukraine 6Department of Surgery and Cancer, Imperial College London, Hammersmith Hospital, London W12 0HS, UK (Correspondence should be addressed to H Zitzelsberger; Email: [email protected]) *(K-J Heiliger and J Hess contributed equally to this work) Abstract For an identification of novel candidate genes in thyroid tumourigenesis, we have investigated gene copy number changes in a Trk-T1 transgenic mouse model of thyroid neoplasia. For this aim, 30 thyroid tumours from Trk-T1 transgenics were investigated by comparative genomic hybridisation. Recurrent gene copy number alterations were identified and genes located in the altered chromosomal regions were analysed by Gene Ontology term enrichment analysis in order to reveal gene functions potentially associated with thyroid tumourigenesis. In thyroid neoplasms from Trk-T1 mice, a recurrent gain on chromosomal bands 1C4–E2.3 (10.0% of cases), and losses on 3H1–H3 (13.3%), 4D2.3–E2 (43.3%) and 14E4–E5 (6.7%) were identified. -

Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways
biomolecules Article Whole Genome Sequencing of Familial Non-Medullary Thyroid Cancer Identifies Germline Alterations in MAPK/ERK and PI3K/AKT Signaling Pathways Aayushi Srivastava 1,2,3,4 , Abhishek Kumar 1,5,6 , Sara Giangiobbe 1, Elena Bonora 7, Kari Hemminki 1, Asta Försti 1,2,3 and Obul Reddy Bandapalli 1,2,3,* 1 Division of Molecular Genetic Epidemiology, German Cancer Research Center (DKFZ), D-69120 Heidelberg, Germany; [email protected] (A.S.); [email protected] (A.K.); [email protected] (S.G.); [email protected] (K.H.); [email protected] (A.F.) 2 Hopp Children’s Cancer Center (KiTZ), D-69120 Heidelberg, Germany 3 Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ), German Cancer Consortium (DKTK), D-69120 Heidelberg, Germany 4 Medical Faculty, Heidelberg University, D-69120 Heidelberg, Germany 5 Institute of Bioinformatics, International Technology Park, Bangalore 560066, India 6 Manipal Academy of Higher Education (MAHE), Manipal, Karnataka 576104, India 7 S.Orsola-Malphigi Hospital, Unit of Medical Genetics, 40138 Bologna, Italy; [email protected] * Correspondence: [email protected]; Tel.: +49-6221-42-1709 Received: 29 August 2019; Accepted: 10 October 2019; Published: 13 October 2019 Abstract: Evidence of familial inheritance in non-medullary thyroid cancer (NMTC) has accumulated over the last few decades. However, known variants account for a very small percentage of the genetic burden. Here, we focused on the identification of common pathways and networks enriched in NMTC families to better understand its pathogenesis with the final aim of identifying one novel high/moderate-penetrance germline predisposition variant segregating with the disease in each studied family. -

TAF10 Complex Provides Evidence for Nuclear Holo&Ndash;TFIID Assembly from Preform
ARTICLE Received 13 Aug 2014 | Accepted 2 Dec 2014 | Published 14 Jan 2015 DOI: 10.1038/ncomms7011 OPEN Cytoplasmic TAF2–TAF8–TAF10 complex provides evidence for nuclear holo–TFIID assembly from preformed submodules Simon Trowitzsch1,2, Cristina Viola1,2, Elisabeth Scheer3, Sascha Conic3, Virginie Chavant4, Marjorie Fournier3, Gabor Papai5, Ima-Obong Ebong6, Christiane Schaffitzel1,2, Juan Zou7, Matthias Haffke1,2, Juri Rappsilber7,8, Carol V. Robinson6, Patrick Schultz5, Laszlo Tora3 & Imre Berger1,2,9 General transcription factor TFIID is a cornerstone of RNA polymerase II transcription initiation in eukaryotic cells. How human TFIID—a megadalton-sized multiprotein complex composed of the TATA-binding protein (TBP) and 13 TBP-associated factors (TAFs)— assembles into a functional transcription factor is poorly understood. Here we describe a heterotrimeric TFIID subcomplex consisting of the TAF2, TAF8 and TAF10 proteins, which assembles in the cytoplasm. Using native mass spectrometry, we define the interactions between the TAFs and uncover a central role for TAF8 in nucleating the complex. X-ray crystallography reveals a non-canonical arrangement of the TAF8–TAF10 histone fold domains. TAF2 binds to multiple motifs within the TAF8 C-terminal region, and these interactions dictate TAF2 incorporation into a core–TFIID complex that exists in the nucleus. Our results provide evidence for a stepwise assembly pathway of nuclear holo–TFIID, regulated by nuclear import of preformed cytoplasmic submodules. 1 European Molecular Biology Laboratory, Grenoble Outstation, 6 rue Jules Horowitz, 38042 Grenoble, France. 2 Unit for Virus Host-Cell Interactions, University Grenoble Alpes-EMBL-CNRS, 6 rue Jules Horowitz, 38042 Grenoble, France. 3 Cellular Signaling and Nuclear Dynamics Program, Institut de Ge´ne´tique et de Biologie Mole´culaire et Cellulaire, UMR 7104, INSERM U964, 1 rue Laurent Fries, 67404 Illkirch, France. -

Structure and Mechanism of the RNA Polymerase II Transcription Machinery
Downloaded from genesdev.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press REVIEW Structure and mechanism of the RNA polymerase II transcription machinery Allison C. Schier and Dylan J. Taatjes Department of Biochemistry, University of Colorado, Boulder, Colorado 80303, USA RNA polymerase II (Pol II) transcribes all protein-coding ingly high resolution, which has rapidly advanced under- genes and many noncoding RNAs in eukaryotic genomes. standing of the molecular basis of Pol II transcription. Although Pol II is a complex, 12-subunit enzyme, it lacks Structural biology continues to transform our under- the ability to initiate transcription and cannot consistent- standing of complex biological processes because it allows ly transcribe through long DNA sequences. To execute visualization of proteins and protein complexes at or near these essential functions, an array of proteins and protein atomic-level resolution. Combined with mutagenesis and complexes interact with Pol II to regulate its activity. In functional assays, structural data can at once establish this review, we detail the structure and mechanism of how enzymes function, justify genetic links to human dis- over a dozen factors that govern Pol II initiation (e.g., ease, and drive drug discovery. In the past few decades, TFIID, TFIIH, and Mediator), pausing, and elongation workhorse techniques such as NMR and X-ray crystallog- (e.g., DSIF, NELF, PAF, and P-TEFb). The structural basis raphy have been complemented by cryoEM, cross-linking for Pol II transcription regulation has advanced rapidly mass spectrometry (CXMS), and other methods. Recent in the past decade, largely due to technological innova- improvements in data collection and imaging technolo- tions in cryoelectron microscopy. -

Supplementary Tables Supplementary Table S1. Treatment Protocols
Supplementary Tables Supplementary Table S1. Treatment protocols pediatric AML patients. Number of Treatment patients ADE-based regimen 64 ADE with bortezomib 14 ADE with mylotarg 6 APL-like treatment 2 TMD-like treatment 1 Relapse therapy 2 Misc treatment/novel agents 5 Unknown 2 Supplemental Table S2: Patient characteristics for the 73 pediatric acute lymphoblastic leukemia patients Characteristics N, % Number of cases 73 ALL subtype Pre-B ALL 57 (78) T-ALL 16 (22) Age, y, median (range) 7.3 (0.2-18.0) Gender Male 40 (55) Female 33 (45) Declared Ethnicity Caucasian 61 (84) Hispanic 44 (72) Non-Hispanic 17 (28) Black American 6 (8) Asian 4 (5) Mixed 2 (3) Hispanic 1 (1) ‘‘SNP’’ Ethnicity European 9 (12) African 6 (8) American Indian 38 (52) Asian 1 (1) Not done 19 (26) Cytogenetics Favorable 15 (21) Intermediate 42 (58) Unfavorable 15 (21) Unknown 1 (1) Risk Group Low Risk 4 (5) Standard/ Intermediate Risk 29 (40) High/Very High Risk 40 (55) CNS status CNS-1 46 (63) CNS-2 20 (27) CNS-3 6 (8) Unknown 2 (3) Response Complete remission 67 (92) Resistant 4 (5) Fail 2 (3) Alive 63 (86) Supplementary Table S3. The table shows the ‘‘Rosetta Stone’’ of the antibody and protein nomenclature, and the R2 for the antibody validation and the primary and secondary antibody dilutions. Secondary, all antibodies that were used in combination with information about the manufacturer of each antibody, the antibody source, and catalog number are listed. Functional Protein Name Rosetta Stone RPPA Staining Details Category Functional Effect Common Name RPPA Antibody Huge Name (added MiMI Antibody R2, WB vs. -
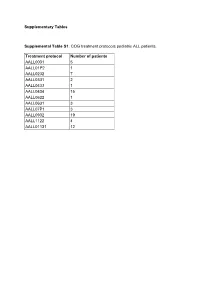
Supplementary Tables Supplemental Table S1. COG Treatment Protocols
Supplementary Tables Supplemental Table S1. COG treatment protocols pediatric ALL patients. Treatment protocol Number of patients AALL0031 5 AALL01P2 1 AALL0232 7 AALL0331 2 AALL0433 1 AALL0434 15 AALL0622 1 AALL0631 3 AALL07P1 3 AALL0932 19 AALL1122 4 AALL01131 12 Supplementary Table S2. This table contains the “Rosetta Stone” for the antibody and protein nomenclature (HUGO, MiMI, GeneCards) together with the RPPA staining details, including the primary and secondary antibody dilutions, the R2 scores For antibody validation, catalog number and the antibody source. Functional Protein Name Rosetta Stone RPPA Staining Details Category Functional Effect Common Name RPPA Antibody Huge Name (added MiMI Antibody R2, WB vs. Antibody 2nd Ab Full Name/Description from GeneCards of Mfg Claimed Manufacturer Catalog# Functional Group Name with PTMs) Name Source RPPA Dilution dilution Phosphorylation Target AKT1 AKT1 AKT1 v-akt murine thymoma viral oncogene homolog 1 AKT1 Cell Signaling 9272 Rabbit > 0.7 200 15000 PI3KAKT AKT1/AKT2/AKT3 AKT1_2_3.pS473 AKT1 v-akt murine thymoma viral oncogene homolog 1 Activation AKT-P473(Ser) Cell Signaling 9271 Rabbit > 0.7 150 20000 PI3KAKT Phospho Ser473 AKT1/AKT2/AKT3 AKT1_2_3.pT308 AKT1 v-akt murine thymoma viral oncogene homolog 1 Activation AKT-P308(Thr) Cell Signaling 9275 Rabbit > 0.7 200 20000 PI3KAKT Phospho Thr308 ASNS ASNS ASNS asparagine synthetase (glutamine-hydrolyzing) ASNS Sigma HPA029318 Rabbit 0.5-0.7 800 20000 Metabolic ATF3 ATF3 ATF3 activating transcription factor 3 ATF3 Abcam ab87213 Rabbit -

TAF4 Takes Flight
COMMENTARY TAF4 takes flight Michael T. Marr II1 Department of Biology and Rosenstiel Basic Medical Sciences Research Center, Brandeis University, Waltham, MA 02454 he eukaryotic mRNA transcrip- from yeast to man, but the region of ETO-TAFH domain was required for tion machinery is exceedingly conservation is limited to a histone H2A activation of the wingless target gene complex, perhaps reflecting the homology region at the carboxyl termi- naked cuticle (nkd) both in cultured requirement for response to nus of the protein (Fig. 1A). The meta- cells and in larval imaginal discs. They Tdiverse environmental and developmen- zoan homologues of TAF4 contain an further showed that this domain directly tal signals. Some of the machinery is extended amino terminus with two addi- interacts with the amino terminus of the common to all mRNA genes and in- tional conserved regions: a glutamine- pygopus, a dedicated transcription factor cludes RNA polymerase II (Pol II) and rich region and a more recently defined for wingless signaling. Interestingly, the a set of general transcription factors ETO-TAFH domain. amino terminus of pygopus has previ- (GTFs) (1). Signal-specific gene expres- The glutamine-rich region was one of ously been defined as absolutely neces- sion patterns are defined by sequence- the first coactivator domains of TFIID sary for Wg/Wnt signaling (14). These specific DNA-binding proteins (activa- to be defined (8). It interacts with a findings place TAF4 at the end of a tors) that bind cognate sites in the number of activators including Sp1 and chain of protein–protein interactions promoter and enhancers of target genes. -

NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer
Florida International University FIU Digital Commons FIU Electronic Theses and Dissertations University Graduate School 11-7-2018 Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer Jairo Ramos [email protected] Follow this and additional works at: https://digitalcommons.fiu.edu/etd Part of the Clinical Epidemiology Commons Recommended Citation Ramos, Jairo, "Decipher Mechanisms by which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer" (2018). FIU Electronic Theses and Dissertations. 3872. https://digitalcommons.fiu.edu/etd/3872 This work is brought to you for free and open access by the University Graduate School at FIU Digital Commons. It has been accepted for inclusion in FIU Electronic Theses and Dissertations by an authorized administrator of FIU Digital Commons. For more information, please contact [email protected]. FLORIDA INTERNATIONAL UNIVERSITY Miami, Florida DECIPHER MECHANISMS BY WHICH NUCLEAR RESPIRATORY FACTOR ONE (NRF1) COORDINATES CHANGES IN THE TRANSCRIPTIONAL AND CHROMATIN LANDSCAPE AFFECTING DEVELOPMENT AND PROGRESSION OF INVASIVE BREAST CANCER A dissertation submitted in partial fulfillment of the requirements for the degree of DOCTOR OF PHILOSOPHY in PUBLIC HEALTH by Jairo Ramos 2018 To: Dean Tomás R. Guilarte Robert Stempel College of Public Health and Social Work This dissertation, Written by Jairo Ramos, and entitled Decipher Mechanisms by Which Nuclear Respiratory Factor One (NRF1) Coordinates Changes in the Transcriptional and Chromatin Landscape Affecting Development and Progression of Invasive Breast Cancer, having been approved in respect to style and intellectual content, is referred to you for judgment. -

Short- and Long-Term Impact of Hyperoxia on the Blood and Retinal Cells’ Transcriptome in a Mouse Model of Oxygen- Induced Retinopathy
www.nature.com/pr BASIC SCIENCE ARTICLE OPEN Short- and long-term impact of hyperoxia on the blood and retinal cells’ transcriptome in a mouse model of oxygen- induced retinopathy Magdalena Zasada1, Anna Madetko-Talowska2, Cecilie Revhaug3,4, Anne Gro W. Rognlien3,4, Lars O. Baumbusch3,Teofila Książek2, Katarzyna Szewczyk2, Agnieszka Grabowska2, Miroslaw Bik-Multanowski2, Jacek Józef Pietrzyk1, Przemko Kwinta1 and Ola Didrik Saugstad3,4 BACKGROUND: We aimed to identify global blood and retinal gene expression patterns in murine oxygen-induced retinopathy (OIR), a common model of retinopathy of prematurity, which may allow better understanding of the pathogenesis of this severe ocular prematurity complication and identification of potential blood biomarkers. METHODS: A total of 120 C57BL/6J mice were randomly divided into an OIR group, in which 7-day-old pups were maintained in 75% oxygen for 5 days, or a control group. RNA was extracted from the whole-blood mononuclear cells and retinal cells on days 12, 17, and 28. Gene expression in the RNA samples was evaluated with mouse gene expression microarrays. RESULTS: There were 38, 1370 and 111 genes, the expression of which differed between the OIR and control retinas on days 12, 17, and 28, respectively. Gene expression in the blood mononuclear cells was significantly altered only on day 17. Deptor and Nol4 genes showed reduced expression both in the blood and retinal cells on day 17. 1234567890();,: CONCLUSION: There are sustained marked changes in the global pattern of gene expression in the OIR mice retinas. An altered expression of Deptor and Nol4 genes in the blood mononuclear cells requires further investigation as they may indicate retinal neovascularization.