Myosin-II Mediated Traction Forces Evoke Localized Piezo1 Ca2+ Flickers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Genetic Variants of PIEZO1 Associate with COVID-19 Fatality
medRxiv preprint doi: https://doi.org/10.1101/2020.06.01.20119651; this version posted September 18, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY 4.0 International license . 1 Genetic variants of PIEZO1 associate with COVID-19 fatality Cheng, C.W., Deivasikamani, V., Ludlow, M.J., De Vecchis, D., Kalli, A.C., Beech, D.J., Sukumar, P. School of Medicine, University of Leeds, Leeds, LS2 9JT, UK. *Authors for correspondence: Dr Piruthivi Sukumar (Lead Author) and Professor David J Beech (Senior Author), Leeds Institute of Cardiovascular and Metabolic Medicine, School of Medicine, LIGHT Laboratories, Clarendon Way, University of Leeds, Leeds, LS2 9JT, United Kingdom. Emails: [email protected]; [email protected]. ABSTRACT Fatality from coronavirus disease 19 (COVID-19) is a major problem globally and so identification of its underlying molecular mechanisms would be helpful. The combination of COVID-19 clinical data and genome sequence information is providing a potential route to such mechanisms. Here we took a candidate gene approach to UK Biobank data based on the suggested roles of endothelium and membrane proteins in COVID-19. We focussed on the PIEZO1 gene, which encodes a non-selective cation channel that mediates endothelial responses to blood flow. The analysis suggests 3 missense PIEZO1 single nucleotide polymorphisms (SNPs) associated with COVID-19 fatality independently of risk factors. All of them affect amino acids in the proximal N-terminus of PIEZO1, which is an unexplored region of the protein. -

S41467-021-21178-4.Pdf
ARTICLE https://doi.org/10.1038/s41467-021-21178-4 OPEN The mechanosensitive Piezo1 channel mediates heart mechano-chemo transduction Fan Jiang1, Kunlun Yin2, Kun Wu 1,5, Mingmin Zhang1, Shiqiang Wang3, Heping Cheng 4, Zhou Zhou2 & ✉ Bailong Xiao 1 The beating heart possesses the intrinsic ability to adapt cardiac output to changes in mechanical load. The century-old Frank–Starling law and Anrep effect have documented that 1234567890():,; stretching the heart during diastolic filling increases its contractile force. However, the molecular mechanotransduction mechanism and its impact on cardiac health and disease remain elusive. Here we show that the mechanically activated Piezo1 channel converts mechanical stretch of cardiomyocytes into Ca2+ and reactive oxygen species (ROS) sig- naling, which critically determines the mechanical activity of the heart. Either cardiac-specific knockout or overexpression of Piezo1 in mice results in defective Ca2+ and ROS signaling and the development of cardiomyopathy, demonstrating a homeostatic role of Piezo1. Piezo1 is pathologically upregulated in both mouse and human diseased hearts via an autonomic response of cardiomyocytes. Thus, Piezo1 serves as a key cardiac mechanotransducer for initiating mechano-chemo transduction and consequently maintaining normal heart function, and might represent a novel therapeutic target for treating human heart diseases. 1 State Key Laboratory of Membrane Biology, Tsinghua-Peking Center for Life Sciences, Beijing Advanced Innovation Center for Structural Biology, IDG/ McGovern Institute for Brain Research, School of Pharmaceutical Sciences, Tsinghua University, Beijing 100084, China. 2 State Key Laboratory of Cardiovascular Disease, Beijing Key Laboratory for Molecular Diagnostics of Cardiovascular Diseases, Center of Laboratory Medicine, Fuwai Hospital, National Center for Cardiovascular Diseases, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing 100037, China. -

Pharmacological Activation of PIEZO1 in Human Red Blood Cells Prevents Plasmodium Falciparum Invasion
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.28.446171; this version posted May 28, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Pharmacological activation of PIEZO1 in human red blood cells prevents Plasmodium falciparum invasion Rakhee Lohia1, Jordy Le Guet1, Laurence Berry1, Hélène Guizouarn2, Roberto Bernal3, Rachel Cerdan1, Manouk Abkarian4, Dominique Douguet5, Eric Honoré5*, Kai Wengelnik6* 1 LPHI, University of Montpellier, CNRS UMR5235, Montpellier, France 2 iBV, University Côte d’Azur, CNRS UMR7277, INSERM U1091, Nice, France 3 Departamento de Física, SMAT-C, Universidad de Santiago de Chile, Santiago, Chile 4 Centre de Biologie Structurale, CNRS UMR5048—INSERM U1054, University of Montpellier, Montpellier, France 5 IPMC, University Côte d’Azur, CNRS, INSERM, UMR7275, Labex ICST, Valbonne, France. 6 LPHI, University of Montpellier, CNRS UMR5235, INSERM, Montpellier, France * Correspondence to: [email protected]; [email protected] Abstract An inherited gain-of–function variant (E756 del) in the mechanosensitive cationic channel PIEZO1 was recently shown to confer a significant protection against severe malaria. Here, we demonstrate in vitro that human red blood cell (RBC) infection by Plasmodium falciparum is prevented by the pharmacological activation of PIEZO1. The PIEZO1 activator Yoda1 inhibits RBC invasion, without affecting parasite intraerythrocytic growth, division or egress. RBC dehydration, echinocytosis and intracellular Na+/K+ imbalance are unrelated to the mechanism of protection. Inhibition of invasion is maintained, even after a prolonged wash out of Yoda1. -

Infection Augments Expression of Mechanosensing Piezo1 Channels in Amyloid Plaque-Reactive Astrocytes
fnagi-10-00332 October 17, 2018 Time: 13:57 # 1 ORIGINAL RESEARCH published: 22 October 2018 doi: 10.3389/fnagi.2018.00332 Infection Augments Expression of Mechanosensing Piezo1 Channels in Amyloid Plaque-Reactive Astrocytes María Velasco-Estevez1,2, Myrthe Mampay1, Hervé Boutin3, Aisling Chaney3,4, Peter Warn5, Andrew Sharp5, Ellie Burgess5, Emad Moeendarbary6,7*, Kumlesh K. Dev2* and Graham K. Sheridan1* 1 Neuroimmulology & Neurotherapeutics Laboratory, School of Pharmacy and Biomolecular Sciences, University of Brighton, Brighton, United Kingdom, 2 Drug Development, Department of Physiology, School of Medicine, Trinity College Dublin, Dublin, Ireland, 3 Wolfson Molecular Imaging Centre, Faculty of Biology, Medicine and Health and Manchester Academic Health Sciences Centre, The University of Manchester, Manchester, United Kingdom, 4 Department of Radiology, Stanford University, Stanford, CA, United States, 5 Evotec (UK) Ltd., Manchester Science Park, Manchester, United Kingdom, 6 Department of Biological Engineering, Massachusetts Institute of Technology, Cambridge, MA, United States, 7 Department of Mechanical Engineering, University College London, London, United Kingdom A defining pathophysiological hallmark of Alzheimer’s disease (AD) is the amyloid plaque; Edited by: an extracellular deposit of aggregated fibrillar Ab1−42 peptides. Amyloid plaques are Alberto Javier Ramos, hard, brittle structures scattered throughout the hippocampus and cerebral cortex Consejo Nacional de Investigaciones and are thought to cause hyperphosphorylation of tau, neurofibrillary tangles, and Científicas y Técnicas (CONICET), Argentina progressive neurodegeneration. Reactive astrocytes and microglia envelop the exterior Reviewed by: of amyloid plaques and infiltrate their inner core. Glia are highly mechanosensitive cells Jingwen Niu, and can almost certainly sense the mismatch between the normally soft mechanical Temple University, United States Vladimir Parpura, environment of the brain and very stiff amyloid plaques via mechanosensing ion The University of Alabama channels. -
![The Function and Regulation of Piezo Ion Channels Jason Wu,1 Amanda[13 TD$IF]H](https://docslib.b-cdn.net/cover/2099/the-function-and-regulation-of-piezo-ion-channels-jason-wu-1-amanda-13-td-if-h-1182099.webp)
The Function and Regulation of Piezo Ion Channels Jason Wu,1 Amanda[13 TD$IF]H
TIBS 1305 No. of Pages 15 Series: Fresh Perspectives from Emerging Experts Review Touch, Tension, and Transduction – The Function and Regulation of Piezo Ion Channels Jason Wu,1 Amanda[13_TD$IF]H. Lewis,1 and Jörg Grandl1,* In 2010, two proteins, Piezo1 and Piezo2, were identified as the long-sought Trends molecular carriers of an excitatory mechanically activated current found in many Piezo proteins were identified in 2010 cells. This discovery has opened the floodgates for studying a vast number of as the pore-forming subunits of excita- mechanotransduction processes. Over the past 6 years, groundbreaking tory mechanosensitive ion channels. research has identified Piezos as ion channels that sense light touch, proprio- Piezo ion channels play essential roles ception, and vascular blood flow, ruled out roles for Piezos in several other in diverse physiological processes ran- mechanotransduction processes, and revealed the basic structural and func- ging from regulation of red blood cell fi volume to sensation of gentle touch, tional properties of the channel. Here, we review these ndings and discuss the and are associated with a number of many aspects of Piezo function that remain mysterious, including how Piezos diseases. convert a variety of mechanical stimuli into channel activation and subsequent A recent medium-resolution structure inactivation, and what molecules and mechanisms modulate Piezo function. gives insight into the overall architec- ture of Piezo1, but does not give straight answers as to how the channel Piezo Proteins: True Mechanically Activated Ion Channels? transduces mechanical force into pore Piezo proteins are pore-forming subunits of ion channels that open in response to mechanical opening. -

Piezo1 Incorporates Mechanical Force Signals Into the Genetic Program That Governs Lymphatic Valve Development and Maintenance
Piezo1 incorporates mechanical force signals into the genetic program that governs lymphatic valve development and maintenance Dongwon Choi, … , Il-Taeg Cho, Young-Kwon Hong JCI Insight. 2019;4(5):e125068. https://doi.org/10.1172/jci.insight.125068. Research Article Vascular biology The lymphatic system plays crucial roles in tissue homeostasis, lipid absorption, and immune cell trafficking. Although lymphatic valves ensure unidirectional lymph flows, the flow itself controls lymphatic valve formation. Here, we demonstrate that a mechanically activated ion channel Piezo1 senses oscillating shear stress (OSS) and incorporates the signal into the genetic program controlling lymphatic valve development and maintenance. Time-controlled deletion of Piezo1 using a pan-endothelial Cre driver (Cdh5[PAC]-CreERT2) or lymphatic-specific Cre driver (Prox1-CreERT2) equally inhibited lymphatic valve formation in newborn mice. Furthermore, Piezo1 deletion in adult lymphatics caused substantial lymphatic valve degeneration. Piezo1 knockdown in cultured lymphatic endothelial cells (LECs) largely abrogated the OSS-induced upregulation of the lymphatic valve signature genes. Conversely, ectopic Piezo1 overexpression upregulated the lymphatic valve genes in the absence of OSS. Remarkably, activation of Piezo1 using chemical agonist Yoda1 not only accelerated lymphatic valve formation in animals, but also triggered upregulation of some lymphatic valve genes in cultured LECs without exposure to OSS. In summary, our studies together demonstrate that Piezo1 is the force sensor in the mechanotransduction pathway controlling lymphatic valve development and maintenance, and Piezo1 activation is a potentially novel therapeutic strategy for congenital and surgery-associated lymphedema. Find the latest version: https://jci.me/125068/pdf RESEARCH ARTICLE Piezo1 incorporates mechanical force signals into the genetic program that governs lymphatic valve development and maintenance Dongwon Choi,1,2 Eunkyung Park,1,2 Eunson Jung,1,2 Boksik Cha,3 Somin Lee,4 James Yu,4 Paul M. -

1 Piezo2 Mechanosensitive Ion Channel Is Located to Sensory
bioRxiv preprint doi: https://doi.org/10.1101/2021.01.20.427483; this version posted January 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Piezo2 mechanosensitive ion channel is located to sensory neurons and non-neuronal cells in rat peripheral sensory pathway: implications in pain Seung Min Shin1, Francie Moehring2, Brandon Itson-Zoske1, Fan Fan3, Cheryl L. Stucky2, Quinn H. Hogan1, 4, and Hongwei Yu1, 4* 1. Department of Anesthesiology, Medical College of Wisconsin, Milwaukee, WI 53226 2. Department of Cell Biology, Neurobiology and Anatomy, Medical College of Wisconsin, Milwaukee, WI 53226 3. Department of Pharmacology and Toxicology, Mississippi University Medical Center, Jackson, Mississippi 39216 4. Zablocki Veterans Affairs Medical Center, Milwaukee, Wisconsin 53295 * Correspondence author: [email protected], phone: 414-955-5745 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.01.20.427483; this version posted January 21, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Piezo2 mechanotransduction channel is a crucial mediator of sensory neurons for sensing and transducing touch, vibration, and proprioception. We here characterized Piezo2 expression and cell specificity in rat peripheral sensory pathway using a validated Piezo2 antibody. Immunohistochemistry using this antibody revealed Piezo2 expression in pan primary sensory neurons (PSNs) of dorsal rood ganglia (DRG) in naïve rats, which was actively transported along afferent axons to both central presynaptic terminals innervating the spinal dorsal horn (DH) and peripheral afferent terminals in skin. -

Piezo1 Regulates Mechanotransductive Release of ATP from Human Rbcs
Piezo1 regulates mechanotransductive release of ATP from human RBCs Eyup Cinara,1, Sitong Zhoua,1, James DeCourceyb, Yixuan Wanga,c, Richard E. Waughd, and Jiandi Wana,2 aMicrosystems Engineering, Rochester Institute of Technology, Rochester, NY 14623; bDepartment of Biology, Rochester Institute of Technology, Rochester, NY 14623; cSchool of Mechanical Engineering, University of Science and Technology, Beijing 100083, China; and dDepartment of Biomedical Engineering, University of Rochester, Rochester, NY 14627 Edited by Nancy E. Kleckner, Harvard University, Cambridge, MA, and approved August 19, 2015 (received for review April 16, 2015) Piezo proteins (Piezo1 and Piezo2) are recently identified mechani- RBCs (9, 17). Pannexin-1, however, is a channel-forming protein cally activated cation channels in eukaryotic cells and associated with and has been suggested as a mechanosensing ATP release channel physiological responses to touch, pressure, and stretch. In particular, (18). Under osmotic stress, for example, ATP released from RBCs human RBCs express Piezo1 on their membranes, and mutations of was attenuated by carbenoxolone, a highly effective pannexin Piezo1 have been linked to hereditary xerocytosis. To date, however, channel blocker, suggesting that pannexin-1 might be one of the physiological functions of Piezo1 on normal RBCs remain poorly un- conductance channels responsible for the mechanotransductive derstood. Here, we show that Piezo1 regulates mechanotransductive release of ATP (18). Although progress has been made in un- release of ATP from human RBCs by controlling the shear-induced derstanding mechanotransductive ATP release from RBCs, many 2+ calcium (Ca ) influx. We find that, in human RBCs treated with questions remain about the signal transduction pathways. For Piezo1 inhibitors or having mutant Piezo1 channels, the amounts of + example, how does mechanical force transduce signals to ATP shear-induced ATP release and Ca2 influx decrease significantly. -

Stimulation of Piezo1 by Mechanical Signals Promotes Bone Anabolism
RESEARCH ARTICLE Stimulation of Piezo1 by mechanical signals promotes bone anabolism Xuehua Li1,2, Li Han3, Intawat Nookaew1,4, Erin Mannen1,2, Matthew J Silva5, Maria Almeida1,2,3, Jinhu Xiong1,2* 1Center for Musculoskeletal Disease Research, University of Arkansas for Medical Sciences, Little Rock, United States; 2Department of Orthopaedic Surgery, University of Arkansas for Medical Sciences, Little Rock, United States; 3Division of Endocrinology, University of Arkansas for Medical Sciences, Little Rock, United States; 4Department of Biomedical Informatics, University of Arkansas for Medical Sciences, Little Rock, United States; 5Department of Orthopaedic Surgery, Washington University, St Louis, United States Abstract Mechanical loading, such as caused by exercise, stimulates bone formation by osteoblasts and increases bone strength, but the mechanisms are poorly understood. Osteocytes reside in bone matrix, sense changes in mechanical load, and produce signals that alter bone formation by osteoblasts. We report that the ion channel Piezo1 is required for changes in gene expression induced by fluid shear stress in cultured osteocytes and stimulation of Piezo1 by a small molecule agonist is sufficient to replicate the effects of fluid flow on osteocytes. Conditional deletion of Piezo1 in osteoblasts and osteocytes notably reduced bone mass and strength in mice. Conversely, administration of a Piezo1 agonist to adult mice increased bone mass, mimicking the effects of mechanical loading. These results demonstrate that Piezo1 is a mechanosensitive ion channel by which osteoblast lineage cells sense and respond to changes in mechanical load and identify a novel target for anabolic bone therapy. DOI: https://doi.org/10.7554/eLife.49631.001 *For correspondence: [email protected] Introduction Competing interests: The Mechanical signals play critical roles in bone growth and homeostasis (Turner et al., 2009; authors declare that no Ozcivici et al., 2010). -
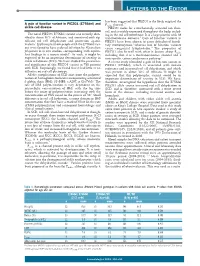
A Gain of Function Variant in PIEZO1 (E756del) and Sickle Cell Disease
LETTERS TO THE EDITOR has been suggested that PIEZO1 is the likely origin of the A gain of function variant in PIEZO1 (E756del) and 4,6 Psickle channel. sickle cell disease PIEZO1 codes for a mechanically activated ion chan - nel, and is widely expressed throughout the body, includ - The novel PIEZO1 E756del variant was recently iden - ing in the red cell membrane. It is a large protein with 36 tified in about 30% of Africans, and associated with sig - transmembrane domains. 8 Gain of function variants in nificant red cell dehydration and protection against PIEZO1 have been shown to cause dehydrated heredi - 1 malaria. Erythrocytes from people with the E756del vari - tary stomatocytosis, 9 whereas loss of function variants ant were found to have reduced infection by Plasmodium cause congenital lymphedema. 10 The properties of falciparum in in vitro studies, corresponding with equiva - PIEZO1 also fit well with what is known about P 1 sickle lent findings in a mouse model. As such, it might be including that it is a mechanosensitive channel and a expected to be an important determinant of severity in non-specific cation conductance pathway. 6 sickle cell disease (SCD). We have studied the prevalence A recent study identified a gain of function variant in and significance of this PIEZO1 variant in 788 patients PIEZO1 (E756del), which is associated with malaria with SCD. Surprisingly, we found that it had little or no resistance and increased red cell dehydration, and which influence on red cell phenotype. was present in about 30% of Africans. 1 It might be All the complications of SCD arise from the polymer - expected that this polymorphic variant would be an ization of hemoglobin molecules incorporating a mutated important determinant of severity in SCD. -

Piezo1 Links Mechanical Forces to Red Blood Cell Volume Stuart M Cahalan1†, Viktor Lukacs1†, Sanjeev S Ranade1, Shu Chien2,3, Michael Bandell4, Ardem Patapoutian1*
SHORT REPORT elifesciences.org Piezo1 links mechanical forces to red blood cell volume Stuart M Cahalan1†, Viktor Lukacs1†, Sanjeev S Ranade1, Shu Chien2,3, Michael Bandell4, Ardem Patapoutian1* 1Department of Molecular and Cellular Neuroscience, Howard Hughes Medical Institute, The Scripps Research Institute, La Jolla, United States; 2Department of Bioengineering, University of California, San Diego, San Diego, United States; 3Institute of Engineering in Medicine, University of California, San Diego, San Diego, United States; 4Genomics Institute of the Novartis Research Foundation, San Diego, United States Abstract Red blood cells (RBCs) experience significant mechanical forces while recirculating, but the consequences of these forces are not fully understood. Recent work has shown that gain-of- function mutations in mechanically activated Piezo1 cation channels are associated with the dehydrating RBC disease xerocytosis, implicating a role of mechanotransduction in RBC volume regulation. However, the mechanisms by which these mutations result in RBC dehydration are unknown. In this study, we show that RBCs exhibit robust calcium entry in response to mechanical stretch and that this entry is dependent on Piezo1 expression. Furthermore, RBCs from blood-cell- specific Piezo1 conditional knockout mice are overhydrated and exhibit increased fragility both in vitro and in vivo. Finally, we show that Yoda1, a chemical activator of Piezo1, causes calcium influx and subsequent dehydration of RBCs via downstream activation of the KCa3.1 Gardos channel, *For correspondence: ardem@ directly implicating Piezo1 signaling in RBC volume control. Therefore, mechanically activated Piezo1 scripps.edu plays an essential role in RBC volume homeostasis. DOI: 10.7554/eLife.07370.001 †These authors contributed equally to this work Competing interests: The authors declare that no Introduction competing interests exist. -

The Mechanosensitive Piezo1 Channel Is Required for Bone Formation
RESEARCH ARTICLE The mechanosensitive Piezo1 channel is required for bone formation Weijia Sun1†, Shaopeng Chi2†, Yuheng Li1, Shukuan Ling1, Yingjun Tan1, Youjia Xu3, Fan Jiang2, Jianwei Li1, Caizhi Liu1, Guohui Zhong1, Dengchao Cao1, Xiaoyan Jin1, Dingsheng Zhao1, Xingcheng Gao1, Zizhong Liu1, Bailong Xiao2*, Yingxian Li1* 1State Key Laboratory of Space Medicine Fundamentals and Application, China Astronaut Research and Training Center, Beijing, China; 2State Key Laboratory of Membrane Biology, Tsinghua-Peking Joint Center for Life Sciences, IDG/McGovern Institute for Brain Research, School of Pharmaceutical Sciences, Tsinghua University, Beijing, China; 3The Second Affiliated Hospital of Soochow University, Suzhou, China Abstract Mechanical load of the skeleton system is essential for the development, growth, and maintenance of bone. However, the molecular mechanism by which mechanical stimuli are converted into osteogenesis and bone formation remains unclear. Here we report that Piezo1, a bona fide mechanotransducer that is critical for various biological processes, plays a critical role in bone formation. Knockout of Piezo1 in osteoblast lineage cells disrupts the osteogenesis of osteoblasts and severely impairs bone structure and strength. Bone loss that is induced by mechanical unloading is blunted in knockout mice. Intriguingly, simulated microgravity treatment reduced the function of osteoblasts by suppressing the expression of Piezo1. Furthermore, *For correspondence: osteoporosis patients show reduced expression of Piezo1, which is closely correlated with [email protected] osteoblast dysfunction. These data collectively suggest that Piezo1 functions as a key (BX); mechanotransducer for conferring mechanosensitivity to osteoblasts and determining mechanical- [email protected] (YL) load-dependent bone formation, and represents a novel therapeutic target for treating †These authors contributed osteoporosis or mechanical unloading-induced severe bone loss.