A Molecular Approach to Calanus (Copepoda:Calanoida) Development and Systematics
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Threatened Birds of the Americas
WHITE-WINGED GUAN Penelope albipennis E1 Discovered in dry north-westernmost Peru in 1876 and generally presumed extinct for a century thereafter, this guan is now known from a small number of dry wooded valleys in the Andean foothills of Peru (chiefly in Lambayeque), where it numbers possibly less than a hundred individuals and is seriously endangered by forest clearance. DISTRIBUTION The White-winged Guan is restricted to a small area of north-west Peru, although the exact extent of its distribution there remains to be clarified; that it might or does occur in adjacent Ecuador is dealt with in the last paragraph in this section. The species was originally described from a specimen collected in December 1876 on Condesa island, 3°31’S 80°29’W, in the Santa Lucia swamps of the Tumbes river delta, Tumbes, and a second specimen was obtained a month later, January 1877, at the Hacienda Pabur, 5°15’S 80°20’W, 200 km to the south and 130 km inland, in Piura, while a third specimen from the same locality is believed to be the live-caught offspring of the second (Taczanowski 1877, 1884-1886, Vaurie 1966b, de Macedo 1978, 1979). At the time of its first discovery, the species was reported at second-hand to inhabit mangroves at the mouth of the Zarumilla (“Zurumilla”) on the border with Ecuador, and to be found in all the larger river valleys of western Peru as far south as the Chicama in La Libertad (but there was no sight record from there, contra Meyer de Schauensee 1966), in particular those at Lambayeque and “Nancho (Rio de Saña)” in adjacent Cajamarca (Taczanowski 1884-1886). -

Forest Stewardship Plan
PUBLIC SUMMARY 2019 FOREST STEWARDSHIP PLAN License number: FSC-C021201 License number: FSC-C008495 ÍNDICE GENERAL INFORMATION 3 INTRODUCTION 4 The Company 4 Mission 4 Vision 5 Principles 5 Land and Occupation and Location 6 WOOD PRODUCTION PROCESS 11 Strategic Planning 12 Long-term Planning (21 years) 12 Tatical Planning - Medium Term 13 Operational Planning - Short Term (18 months) 13 Technical, Economic, Environmental, and Social Planning - TEESP 13 Seedling Production 14 Forest Activities 15 Inventory 15 Harvest/Transportation 15 FORESTRY RESEARCH 16 Genetic Improvement 17 Forest Plague Control and Stewardship 17 Soil, Nourishment, and Forest Stewardship 17 FOREST PROTECTION 19 Forest Fire Prevention and Control 19 Property Protection and Community Access to the Company’s Land 22 ENVIRONMENT 23 Environmental Monitoring 23 Vegetation 24 Wildlife 29 Water Resources 33 High Conservation Value Areas – HCVA 36 Environmental Education and Communication 41 HUMAN RESOURCES POLICY 42 Recruitment, Selection, and Compensation 42 Personnel 43 Occupational Health and Safety 44 Benefits 48 Training, Development, and Quality of Life 48 Socioeconomic profile of CENIBRA’s area of influence 48 SOCIAL ASPECTS – CENIBRA INSTITUTE 51 CENIBRA INSTITUTE’s Mission 52 CENIBRA INSTITUTE’s Principles 52 Strategy 50 Areas of Interest 53 CONTACT 58 2 Public Summary of the Forest Stewardship Plan GENERAL INFORMATION Name: Celulose Nipo-Brasileira S.A. - CENIBRA Address: BR-381, km 172 - Belo Oriente - Minas Gerais - CEP: 35196-972 Phone/Fax: (31) 3829 5010 / 3829 5260 E-mail: [email protected] Internet: www.cenibra.com.br State Registation: 063.141486.0136 Taxpayer´s Number (CNPJ/MF): 42.278.796/0001-99 Legal Representative: Júlio César Tôrres Ribeiro / Technical and Industrial Director This document’s version: Version 15, 2019; this is the 15th version after the initial certification in 2005. -

ON 92 Bonanomi Et Al.Fm
(2016) 27: 133–135 ____________________________________________________________________________ ABERRANT PLUMAGE IN THE WHITE‐THROATED PIPING‐GUAN (PIPILE GRAYI PELZELN, 1870, CRACIDAE) ____________________________________________________________________________ Juliana Bonanomi 1 · Fernando Rodrigo Tortato 1,2 · João Basta de Pinho 1,3 1Programa de Pós‐graduação em Ecologia e Conservação da Biodiversidade ‐ Universidade Federal de Mato Grosso, CEP 78060‐900, Cuiabá, MT, Brazil 2Panthera – Pantanal Jaguar Project. Caixa Postal 3203, Cuiabá, Mato Grosso, CEP 78060‐970, Brazil 3Laboratório de Ornitologia, Instituto de Biociências, Universidade Federal de Mato Grosso, CEP 78060‐900, Cuiabá, MT, Brazil E‐mail: Juliana Bonanomi ∙ [email protected] ABSTRACT ∙ There is limited information regarding cases of aberrant plumage in Cracidae. Here we present the first record of aberrant plumage in the White‐throated Piping‐guan (Pipile grayi). The individual with aberrant plumage was predominantly white‐colored, but with pale cream underparts. Unfeathered body parts like the fleshy wattle, bill, and tarsus had pink color. Based on these characteristics we conclude that it likely constitutes a case of “Ino‐light” mutation. RESUMO ∙ Plumagem aberrante em Jacutinga‐de‐barbela‐branca (Pipile grayi Pelzeln, 1870, Cracidae) Informações referentes a aberrações na coloração de plumagem são limitadas. Nesta comunicação breve apresenta‐ mos o primeiro registro de coloração aberrante de plumagem de Jacutinga‐de‐barbela‐branca (Pipile grayi). O indi‐ víduo apresentou plumagem com coloração predominantemente branca, com as partes inferiores com coloração creme. Outras partes do corpo, como barbela, bico e tarso apresentaram coloração rosada. Através destas caracterís‐ ticas fenotípicas e utilizando‐se de chaves de identificação para mutação de plumagem em aves disponíveis na litera‐ tura é suscetível que esta aberração se trate de um caso de mutação “Ino‐light”. -

In Brazil, There Are Seven Species of the Genus Penelope (Cracidae: Galliformes)
ISSN 1981-1268 COSTA & CHRISTOFFERSEN (2016) 173 http://dx.doi.org/10.21707/gs.v10.n04a14 COMPARATIVE ANALYSIS OF VOCALIZATIONS OF GUANS (GALLIFORMES, CRACIDAE, PENELOPE) FROM BRAZIL (SOUTH AMERICA) DIMITRI DE ARAUJO COSTA1 & MARTIN LINDSEY CHRISTOFFERSEN1 1 Universidade Federal da Paraíba. Centro de Ciências Exatas e da Natureza. Departamento de Sistemática e Ecologia. João Pessoa, Paraíba, Brasil. Recebido em 23 de setembro de 2015. Aceito em 11 de julho de 2016. Publicado em 30 de setembro de 2016. ABSTRACT – The genus Penelope is a group of guans of the family Cracidae, Order Galliformes, which is widely distributed in Brazil, totaling seven species (Penelope jacquacu, P. jacucaca, P. marail, P. pileata, P. obscura, P. ochrogaster and P. superciliaris). Each guan species has a particular distribution, with the exception of the Rusty-margined Guan (P. superciliaris) that is found in almost all the national territory. The objective of this study involves analyzing the songs of the species of Penelope in different regions of Brazil, and comparing them to those of the species P. superciliaris to know if their singing patterns are modified when overlapping with other species of the genus, thus avoiding competition by acoustic interference. This comparative analysis involving several guan species is the first performed in cracids in the world, thus being highly relevant for future studies. Furthermore, we are providing the diagnostic characteristics, conservation status and ecological data of each species of the genus Penelope reported from Brazil. KEY WORDS: ACOUSTIC INTERFERENCE; APOMORPHIES; COMPETITION; CONSERVATION STATUS; CRACIDS. ANÁLISE COMPARATIVA DAS VOCALIZAÇÕES DOS JACUS (GALLIFORMES, CRACIDAE, PENELOPE) DO BRASIL (AMÉRICA DO SUL) RESUMO – O gênero Penelope é um grupo de jacus da família Cracidae, Ordem Galliformes, que é amplamente distribuído no Brasil, totalizando sete espécies (Penelope jacquacu, P. -

Penelope Albipennis, with Notes on Its Nest
THE WILSON BULLETIN A QUARTERLYMAGAZINE OF ORNITHOLOGY Published by the Wilson Ornithological Society VOL. 94, No. 3 SEPTEMBER 1982 PAGES 241-432 N’Xwn Bull., 94(3), 1982, pp. 241-259 SYSTEMATIC RELATIONSHIPS AND ZOOGEOGRAPHY OF THE WHITE-WINGED GUAN (PENELOPE ALBIPENlV1S) AND RELATED FORMS J. WILLIAM ELEY The avian genus Penelope (Cracidae), comprising 13-15 species (de- pending on the authority), is widely distributed from northeastern Mexico south to Argentina and Uruguay. Relationships within this genus are ob- scure, as reflected by the variation in recent taxonomic treatments (Vuil- leumier 1965, Vaurie 1968, Delacour and Amadon 1973). There is general agreement by these authors that members of the genus fall into several species groups, although the exact composition and arrangement of these groups are also debated. Vuilleumier (1965) divided the genus into four species groups, and included species of Pipile in Penelope. Vuilleumiers’ (1965) conclusion that there are only eight species of Penelope (including Pipile) was not accepted in two subsequent treatments of the family. Vau- rie (1968) and Delacour and Amadon (1973) generally agreed that the genus is divisible into one group of small species (P. montugnii [Andean Guan], P. argyrotis [Band-tailed Guan], P. barbata [Bearded Guan], P. marail [Marail Guan], P. ortoni [Baud6 Guan], and P. superciliaris [Rusty-mar- gined Guan], and two groups of large-sized species (P. purpuruscens [Crested Guan], P. jucquucu [Spixs’ Guan], P. obscuru [Dusky-legged Guan], P. dubbenei [Red-faced Guan], and P. ulbipennis [White-winged Guan] forming one group; and P. pileutu [White-crested Guan], P. och- roguster [Chestnut-bellied Guan], and P. -
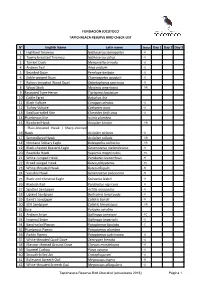
N° English Name Latin Name Status Day 1 Day 2
FUNDACIÓN JOCOTOCO TAPICHALACA RESERVE BIRD CHECK-LIST N° English Name Latin name Status Day 1 Day 2 Day 3 1 Highland Tinamou Nothocercus bonapartei - R 2 Tawny-breasted Tinamou Nothocercus julius - U 3 Torrent Duck Merganetta armata - U 4 Andean Teal Anas andium - U 5 Bearded Guan Penelope barbata - U 6 Sickle-winged Guan Chamaepetes goudotii - U 7 Rufous-breasted Wood Quail Odontophorus speciosus - R 8 Wood Stork Mycteria americana - VR 9 Fasciated Tiger-Heron Tigrisoma fasciatum 10 Cattle Egret Bubulcus ibis 11 Black Vulture Coragyps atratus - U 12 Turkey Vulture Cathartes aura - U 13 Swallow-tailed Kite Elanoides forficatus - U 14 Plumbeous Kite Ictinia plumbea 15 Bicolored Hawk Accipiter bicolor - VR Plain-breasted Hawk / Sharp-shinned 16 Hawk Accipiter striatus - U 17 Semicollared Hawk Accipiter collaris - VR 18 Montane Solitary Eagle Buteogallus solitarius - VR 19 Black-chested Buzzard-Eagle Geranoaetus melanoleucus - R 20 Roadside Hawk Rupornis magnirostris - FC 21 White-rumped Hawk Parabuteo leucorrhous - R 22 Broad-winged Hawk Buteo platypterus - FC 23 White-throated Hawk Buteo albigula - R 24 Variable Hawk Geranoaetus polyosoma - R 25 Black-and-chestnut Eagle Spizaetus isidori - R 26 Blackish Rail Pardirallus nigricans - R 27 Spotted Sandpiper Actitis macularius - R 28 Upland Sandpiper Bartramia longicauda - R 29 Baird's Sandpiper Calidris bairdii - R 30 Stilt Sandpiper Calidris himantopus - VR 31 Sora Porzana carolina 32 Andean Snipe Gallinago jamesoni - FC 33 Imperial Snipe Gallinago imperialis - U 34 Band-tailed Pigeon Patagioenas -

The Rusty-Margined Guan (Penelope Superciliaris) in the Brazilian Atlantic Rain Forest: Density, Population Size, Activity and Habitat Use
CSIRO PUBLISHING Wildlife Research https://doi.org/10.1071/WR17161 The rusty-margined guan (Penelope superciliaris) in the Brazilian Atlantic rain forest: density, population size, activity and habitat use Juliane Pereira-Ribeiro A,D, Átilla Colombo Ferreguetti A, Walfrido Moraes Tomas B, Helena Godoy Bergallo A, Carlos Frederico Duarte RochaA and Daniel M. Brooks C ADepartment of Ecology, Rio de Janeiro State University, Rio de Janeiro, RJ, CEP 20550-019, Brazil. BWildlife Laboratory, Embrapa Pantanal, Mato Grosso do Sul, CEP 79320-900, Brazil. CDepartment. Vertebrate Zoology, Houston Museum of Natural Science, Houston, TX 77030, USA. DCorresponding author. Email: [email protected] Abstract Context. Population estimation and monitoring are important tools for the support of programs and actions of conservation for most wildlife species, including birds. For validation of such support, a set of reliable, consistent and comparable parameter estimates is essential (through systematic monitoring over time), in addition to studies on ecological aspects of the species. Aims. In this study, our aim was to analyse the population of Penelope superciliaris in the Vale Natural Reserve (VNR), located in north-eastern Espírito Santo, Brazil, providing estimates of density, population size and activity. We also analysed occupancy and detectability of the species, relating them to six covariates (distance from forest edge, water and road, as well as poaching intensity, canopy cover and understorey cover) based on previous knowledge of the ecology of rusty-margined guan. Methods. We used line-transect surveys to estimate density and abundance, and camera traps to estimate habitat use by P. superciliaris, using the occupancy modelling approach. Key results. -

On the Status of Dusky-Legged Guan Penelope Obscura Temminick, 1815
Volume 55(14):211‑216, 2015 ON THE STATUS OF DUSKY-LEGGED GUAN PENELOPE OBSCURA TEMMINCK, 1815 (AVES: CRACIDAE) IN PARAGUAY PAUL SMITH1,2 ROBERTO DERNA1 ABSTRACT Despite repeated references to the species in Paraguay, the status of the Dusky-legged Guan Pe- nelope obscura in the country has been the subject of much debate. In an attempt to clarify the available data, a thorough review of literature records is provided and details of new and previ- ously unpublished records that confirm that the nominate subspecies is present in Paraguay are given. With the species limits in the obscura complex poorly defined, we provide a brief discus- sion of the potential importance of Paraguayan populations for the conservation of the species. Key-Words: Conservation; Distribution; Galliformes. INTRODUCTION cords until recently. In this note we provide a detailed summary of previous published Paraguayan records Despite Azara’s (1805) claim that the “Yacuhú” and information about a newly discovered and semi- (upon which Temminck’s description is based) “no es- habituated population of the species in the south of casea en dicha Provincia” (is not scarce in Paraguay), the country. the presence or absence of the Dusky-legged Guan Penelope obscura within the boundaries of modern day Paraguay has long been the subject of debate (Dela- PREVIOUS PUBLISHED RECORDS cour et al., 2004). Hayes (1995) concluded that a lack of details and an absence of verifiable specimen Azara (1805) provides a fairly detailed descrip- records indicated that further documentation was re- tion of his Yacuhú that was used as the basis of Tem- quired before the species could be accepted onto the minck’s (1815) Penelope obscura. -

A Phonological Description of “Pet Talk” in Arara
A PHONOLOGICAL DESCRIPTION OF “PET TALK” IN ARARA by Isaac Costa de Souza Bachelor of Arts, Universidade Federal do Pará, 1980 Master of Arts, Universidade Estadual de Campinas, 1988 A Thesis Submitted to the Graduate Faculty of the University of North Dakota in partial fulfillment of the requirements for the degree of Master of Arts Grand Forks, North Dakota August 2010 This thesis, submitted by Isaac Costa de Souza in partial fulfillment of the requirements for the Degree of Master of Arts from the University of North Dakota, has been read by the Faculty Advisory Committee under whom the work has been done and is hereby approved. ___________________________________ Chair ___________________________________ ___________________________________ This thesis meets the standards for appearance, conforms to the style and format requirements of the Graduate School of the University of North Dakota, and is hereby approved. __________________________________ Dean of the Graduate School __________________________________ Date ii PERMISSION Title A Phonological Description of “Pet Talk” in Arara Department Linguistics Degree Master of Arts In presenting this thesis in partial fulfillment of the requirements for a graduate degree from the University of North Dakota, I agree that the library of this University shall make it freely available for inspection. I further agree that permission for extensive copying for scholarly purposes may be granted by the professor who supervised my thesis work or, in his absence, by the chairperson of the department or the dean of the Graduate School. It is understood that any copying or publication or other use of this thesis or part thereof for financial gain shall not be allowed without my written permission. -

Management of Galliformes
38_Galliforms.qxd 8/24/2005 10:02 AM Page 861 CHAPTER 38 Management of Galliformes GARY D. BUTCHER, BS, MS, DVM, P hD, D ipl ACPV Members of the order Galliformes are found on every continent except Antarctica. The red junglefowl, com- mon turkey and helmeted guinea fowl have been domesticated for centuries. Their descendants, through selective breeding, are of considerable economic impor- tance today. Some varieties are very plentiful in the wild, while others like the Japanese quail (Coturnix japonica) and various pheasants are approaching a level of com- plete domestication. Many Galliformes are commonly maintained as game and food (meat and/or eggs) birds. Some are stable in captivity under variable ambient conditions, easy to breed and inexpensive. Other species are from niches with specific environmental requirements and need spe- cialized diets, humidity and temperature ranges to sur- vive. Currently, commercial production of chickens and turkeys in the USA for food has surpassed that of the Greg J. Harrison Greg J. Fig 38.1 | In warm climates, game cocks are housed on “string beef, pork and fish industries. In 1900, per capita con- walks”. A leg leash, just long enough for the bird to reach a sumption of chicken was 1 pound and had risen to 80 shelter but not to fight with other birds, allows group confine- pounds by the year 2000! ment. Fighting of cocks is considered inhumane and illegal in some countries; however, it is a part of the culture in others. In this chapter, “domestic fowl” means Gallus gallus, forma domestica (domestic form of the red junglefowl); “domestic turkey” is Meleagris gallopavo, forma domes- tica (domestic form of the common turkey) and “domes- tic guinea fowl” is Numida meleagris, forma domestica (domestic form of the helmeted guinea fowl) (Table 38.1). -

Ecology and Behaviour of the Critically Endangered Trinidad Piping-Guan Aburria Pipile
Vol. 6: 223–229, 2009 ENDANGERED SPECIES RESEARCH Printed March 2009 doi: 10.3354/esr00153 Endang Species Res Published online January 2, 2009 OPENPEN ACCESSCCESS Ecology and behaviour of the critically endangered Trinidad piping-guan Aburria pipile Floyd E. Hayes1, 3,*, Clifmond L. Shameerudeen2, 4, Bryan Sanasie2, 5, Brett D. Hayes1, 3, Carol L. Ramjohn1, Floyd B. Lucas1 1Department of Life Sciences, University of the West Indies, St. Augustine, Trinidad and Tobago 2Department of Biology, University of the Southern Caribbean, Maracas Valley, Trinidad and Tobago 3Present address: Department of Biology, Pacific Union College, 1 Angwin Ave., Angwin, California 94508, USA 4Present address: 307 Michigan Street, Niles, Michigan 49120, USA 5Present address: 4867 Greenfield Dr. Apt. 1, Berrien Springs, Michigan 49103, USA ABSTRACT: From 1997 to 2001 we studied a group of 6 Trinidad piping-guans Aburria pipile, a crit- ically endangered species, in northern Trinidad. The group occupied a home range of 19 ha in highly disturbed secondary forest and around small farms, and often split into smaller groups. They spent 93.6% of the time in the forest canopy >5 m above ground and only 0.2% of the time on the ground (n = 48.1 h). In early morning (n = 18.7 h), they spent 80.3% of their time perched alert, 9.3% preen- ing, 3.9% flying, 3.6% walking, 2.4% feeding, 0.3% drinking, and 0.1% perched flapping. In late afternoon (n = 8.2 h), they spent 78.6% of their time perched alert, 12.8% preening, 3.7% walking, 3.0% feeding, 1.2% flying, and 0.7% drinking. -

ECOLOGY and CONSERVATION of the JACUTINGA Pipile Jacutinga in the ATLANTIC FOREST of BRAZIL
Biological Conservation 82 (1997) 31-39 © 1997 Published by Elsevier Science Ltd All rights reserved. Printed in Great Britain PII: S0006-3207(97)00004-9 0006-3207/97 $17.00 + 0.00 ELSEVIER ECOLOGY AND CONSERVATION OF THE JACUTINGA Pipile jacutinga IN THE ATLANTIC FOREST OF BRAZIL Mauro Galetti," Paulo Martuscelli, b Ffibio Olmos c & Alexandre Aleixo d a Wildlife Research Group, Department of Anatomy, University of Cambridge, Cambridge CB2 3D Y, UK bCP194, Peru[be, Sdo Paulo, 11750-970, Sfo Paulo, Brazil ¢Se¢~o de Animais Silvestres, IF, CP 1322, 01509-970, Sdo Paulo, SP, Brazil dDepartamento de Zoologia, UNICAMP, CP 6109, 13083-970 Campinas, Sdo Paulo, Brazil (Received 28 February 1996; revised version received 2 November 1996; accepted 8 November 1996) Abstract from the Neotropics (Silva & Strahl, 1991; Thiollay, The jacutinga Pipile jacutinga was formerly one of the 1994). most abundant game bird cracids in the Atlantic forest of The black-fronted piping-guan or jacutinga Pipile Brazil. Nowadays this species is vulnerable to extinction jacutinga is a medium-sized (up to 1.5 kg) cracid known due to hunting and habitat loss. The ecology of the jacu- from the moist forests of coastal Brazil, the Atlantic tinga was studied at Parque Estadual lntervales, Sdo forest, from southern Bahia to Rio Grande do Sul, and Paulo, Brazil from October 1993 to December 1995 and adjacent areas of Paraguay and Argentina (Delacour & in adjacent areas. Jacutingas were observed to feed Amadon, 1973; Sick, 1993). The species was extremely mainly on the sugar-rich fruit of 41 species. We recorded common in parts of its range until the 1940s and 1950s, a low index of abundance for the jacutinga (0.018) or when thousands could be killed over a short period c.1.7 birds/km 2 at Intervales, one of the best protected (Sick, 1993).