Supporting Information Modular Control of L-Tryptophan Isotopic Substitution Via an Efficient Biosynthetic Cascade
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

National Classification? 13
NATIONAL CL ASSIFICATION INFORMATION FOR MULTI CLASS SWIMMERS Version 1.2 2019 PRINCIPAL PARTNER MAJOR PARTNERS CLASSIFICATION PARTNERS Version 1.2 2019 National Swimming Classification Information for Multi Class Swimmers 1 CONTENTS TERMINOLOGY 3 WHAT IS CLASSIFICATION? 4 WHAT IS THE CLASSIFICATION PATHWAY? 4 WHAT ARE THE ELIGIBLE IMPAIRMENTS? 5 CLASSIFICATION SYSTEMS 6 CLASSIFICATION SYSTEM PARTNERS 6 WHAT IS A SPORT CLASS? 7 HOW IS A SPORT CLASS ALLOCATED TO AN ATHLETE? 7 WHAT ARE THE SPORT CLASSES IN MULTI CLASS SWIMMING? 8 SPORT CLASS STATUS 11 CODES OF EXCEPTION 12 HOW DO I CHECK MY NATIONAL CLASSIFICATION? 13 HOW DO I GET A NATIONAL CLASSIFICATION? 13 MORE INFORMATION 14 CONTACT INFORMATION 16 Version 1.2 2019 National Swimming Classification Information for Multi Class Swimmers 2 TERMINOLOGY Assessment Specific clinical procedure conducted during athlete evaluation processes ATG Australian Transplant Games SIA Sport Inclusion Australia BME Benchmark Event CISD The International Committee of Sports for the Deaf Classification Refers to the system of grouping athletes based on impact of impairment Classification Organisations with a responsibility for administering the swimming classification systems in System Partners Australia Deaflympian Representative at Deaflympic Games DPE Daily Performance Environment DSA Deaf Sports Australia Eligibility Criteria Requirements under which athletes are evaluated for a Sport Class Evaluation Process of determining if an athlete meets eligibility criteria for a Sport Class HI Hearing Impairment ICDS International Committee of Sports for the Deaf II Intellectual Impairment Inas International Federation for Sport for Para-athletes with an Intellectual Disability General term that refers to strategic initiatives that address engagement of targeted population Inclusion groups that typically face disadvantage, including people with disability. -

United States Olympic Committee and U.S. Department of Veterans Affairs
SELECTION STANDARDS United States Olympic Committee and U.S. Department of Veterans Affairs Veteran Monthly Assistance Allowance Program The U.S. Olympic Committee supports Paralympic-eligible military veterans in their efforts to represent the USA at the Paralympic Games and other international sport competitions. Veterans who demonstrate exceptional sport skills and the commitment necessary to pursue elite-level competition are given guidance on securing the training, support, and coaching needed to qualify for Team USA and achieve their Paralympic dreams. Through a partnership between the United States Department of Veterans Affairs and the USOC, the VA National Veterans Sports Programs & Special Events Office provides a monthly assistance allowance for disabled Veterans of the Armed Forces training in a Paralympic sport, as authorized by 38 U.S.C. § 322(d) and section 703 of the Veterans’ Benefits Improvement Act of 2008. Through the program the VA will pay a monthly allowance to a Veteran with a service-connected or non-service-connected disability if the Veteran meets the minimum VA Monthly Assistance Allowance (VMAA) Standard in his/her respective sport and sport class at a recognized competition. Athletes must have established training and competition plans and are responsible for turning in monthly and/or quarterly forms and reports in order to continue receiving the monthly assistance allowance. Additionally, an athlete must be U.S. citizen OR permanent resident to be eligible. Lastly, in order to be eligible for the VMAA athletes must undergo either national or international classification evaluation (and be found Paralympic sport eligible) within six months of being placed on the allowance pay list. -

EG Web Catalog SA-ELCB10
ELECTRICAL SOLUTIONS A. System Overview DOME-TOP ® Barb Ty Clamp Ties – Nylon 6.6 • For indoor use • Stainless steel locking barb provides consistent performance, B1. • Used to secure a cable bundle to another surface such as reliability, and infinite adjustability through entire bundle range Cable Ties a control panel, communication rack, wall or ceiling • Curved tip is easy to pick up from flat surfaces and allows • Design allows for bundling before or after screwing clamp faster initial threading to speed installation in place B2. Cable Accessories B3. Stainless Steel Ties Straight Tip Curved Tip C1. Wiring Duct Nominal Max. Min. C2. Surface Hole Metric Bundle Loop Recommended Std. Std. Raceway Length Width Thickness Dia. Screw Screw Dia. Tensile Str. Installation Pkg. Ctn. Part Number In. mm In. mm In. mm In. mm Size Size In. mm Lbs. N Tool Qty. Qty. C3. Miniature Cross Section Abrasion BC1M-S4-M 4.6 117 .095 2.4 .046 1.2 .122 3.1 #4 M2.5 .90 23 18 80 1000 50000 Protection GTS, GTSL, GS2B, PTS, BC2M-S4-M 8.3 211 .095 2.4 .046 1.2 .122 3.1 #4 M2.5 2.00 51 18 80 1000 25000 PPTS, STS2 C4. Intermediate Cross Section Cable Management BC1.5I-S8-M 6.6 168 .141 3.6 .041 1.0 .174 4.4 #8 M4 1.50 38 40 178 GTS, GTSL, 1000 25000 GS2B, PTS, PPTS, STS2 D1. Standard Cross Section Terminals BC2S-S10-C 8.5 216 .185 4.7 .052 1.3 .200 5.1 #10 M5 2.00 51 50 222 GTS, GTSL, 100 1000 GS2B, GTH, BC3S-S10-D 12.5 318 .185 4.7 .052 1.3 .200 5.1 #10 M5 3.00 76 50 222GS4H, PTS, 500 5000 D2. -

Para Cycling Information Sheet About the Sport Classification Explained
Para cycling information sheet About the sport Para cycling is cycling for people with impairments resulting from a health condition (disability). Para athletes with physical impairments either compete on handcycles, tricycles or bicycles, while those with a visual impairment compete on tandems with a sighted ‘pilot’. Para cycling is divided into track and road events, with seven events in total. Classification explained In Para sport classification provides the structure for fair and equitable competition to ensure that winning is determined by skill, fitness, power, endurance, tactical ability and mental focus – the same factors that account for success in sport for able-bodied athletes. The Para sport classification assessment process identifies the eligibility of each Para athlete’s impairment, and groups them into a sport class according to the degree of activity limitation resulting from their impairment. Classification is sport-specific as an eligible impairment affects a Para athlete’s ability to perform in different sports to a different extent. Each Para sport has a different classification system. Standard Classification in detail Para-Cycling sport classes include: Handcycle sport classes H1 – 5: There are five different sport classes for handcycle racing. The lower numbers indicate a more severe activity limitation. Para athletes competing in the H1 classes have a complete loss of trunk and leg function and limited arm function, e.g. as a result of a spinal cord injury. Para athletes in the H4 class have limited or no leg function, but good trunk and arm function. Para cyclists in sport classes H1 – 4 compete in a reclined position. Para cyclists in the H5 sport class sit on their knees because they are able to use their arms and trunk to accelerate the handcycle. -

(VA) Veteran Monthly Assistance Allowance for Disabled Veterans
Revised May 23, 2019 U.S. Department of Veterans Affairs (VA) Veteran Monthly Assistance Allowance for Disabled Veterans Training in Paralympic and Olympic Sports Program (VMAA) In partnership with the United States Olympic Committee and other Olympic and Paralympic entities within the United States, VA supports eligible service and non-service-connected military Veterans in their efforts to represent the USA at the Paralympic Games, Olympic Games and other international sport competitions. The VA Office of National Veterans Sports Programs & Special Events provides a monthly assistance allowance for disabled Veterans training in Paralympic sports, as well as certain disabled Veterans selected for or competing with the national Olympic Team, as authorized by 38 U.S.C. 322(d) and Section 703 of the Veterans’ Benefits Improvement Act of 2008. Through the program, VA will pay a monthly allowance to a Veteran with either a service-connected or non-service-connected disability if the Veteran meets the minimum military standards or higher (i.e. Emerging Athlete or National Team) in his or her respective Paralympic sport at a recognized competition. In addition to making the VMAA standard, an athlete must also be nationally or internationally classified by his or her respective Paralympic sport federation as eligible for Paralympic competition. VA will also pay a monthly allowance to a Veteran with a service-connected disability rated 30 percent or greater by VA who is selected for a national Olympic Team for any month in which the Veteran is competing in any event sanctioned by the National Governing Bodies of the Olympic Sport in the United State, in accordance with P.L. -

Official Rules
2018 Willamette Valley Youth Football & Cheer OFFICIAL RULES Page 1 Willamette Valley Youth Football & Cheer Table of Contents Part I – The WVYFC Program ............................................................... 5 Article 1: Members Code of Conduct ............................................. 5 Part II – WVYFC Structure .................................................................... 7 Part III – Regulations ............................................................................ 7 Article 1: Authority of League ......................................................... 7 Article 2: Boundaries ....................................................................... 7 Article 3: Coaches Requirements .................................................... 8 Article 4: Registration ..................................................................... 8 Article 5: Formation of Teams ........................................................ 9 Article 6: Mandatory Cuts ............................................................. 10 Article 7: Voluntary Cuts ............................................................... 10 Article 8: Certification ................................................................... 10 Article 9: Retention of Eligibility ................................................... 10 Article 10: No All Stars .................................................................. 10 Article 11: Awards ......................................................................... 11 Article 12: Practice (Definition & Date -

Should the Para-Cycling Classification System Be Reclassified?
Should the para-cycling classification system be reclassified? David Borg1, John Osborne2, Johanna Liljedahl3, Michele Foster1, Carla Nooijen3 1The Hopkins Centre, Menzies Health Institute Queensland, Griffith University, Brisbane, Australia. 2School of Exercise and Nutrition Sciences, Queensland University of Technology, Brisbane, Australia. 3The Swedish School of Sport and Health Sciences (GIH), Stockholm, Sweden. Introduction Results Para-cycling athletes compete on bicycles (C1–5), handcycles The number of men in C1, C2, C3, C4 and C5 was 32, 76, 63, (H1–5) and tricycles (T1-2) in classes based on their functional 76, and 87, respectively. Nineteen men competed in T1 and 58 disability. Higher classes reflect less functional impairment. Para- in T2. The age range of men was 14–62 years. cycling classification is governed by the Union Cycliste Inter- The number of women competing in C1, C2, C3, C4 and C5 was nationale (UCI). The system aims to promote participation in the 4, 18, 16, 20 and 28, respectively. Eleven competed in T1 and sport, by controlling for the impact of impairment on the outcome 15 in T2. Women’s age ranged 17–55 years. of competition. Road race velocity for the bicycling and tricycling is shown in Recently, the separation between adjacent handcycling classes for Figure 1. Comparisons between adjacent classes for men and official UCI events was described––providing benchmarks for women are displayed in Table 2. active and potential athletes. Unfortunately, similar evaluation of the bicycling and tricycling disciplines has not been considered. With the expectation of C4 and C5 for women, the analysis showed that men's and women's road race performance was This study aimed to described the separation between adjacent statistically different between adjacent classes for bicycling and classes, based on performance, in UCI road race events for tricycling. -

The Paralympic Athlete Dedicated to the Memory of Trevor Williams Who Inspired the Editors in 1997 to Write This Book
This page intentionally left blank Handbook of Sports Medicine and Science The Paralympic Athlete Dedicated to the memory of Trevor Williams who inspired the editors in 1997 to write this book. Handbook of Sports Medicine and Science The Paralympic Athlete AN IOC MEDICAL COMMISSION PUBLICATION EDITED BY Yves C. Vanlandewijck PhD, PT Full professor at the Katholieke Universiteit Leuven Faculty of Kinesiology and Rehabilitation Sciences Department of Rehabilitation Sciences Leuven, Belgium Walter R. Thompson PhD Regents Professor Kinesiology and Health (College of Education) Nutrition (College of Health and Human Sciences) Georgia State University Atlanta, GA USA This edition fi rst published 2011 © 2011 International Olympic Committee Blackwell Publishing was acquired by John Wiley & Sons in February 2007. Blackwell’s publishing program has been merged with Wiley’s global Scientifi c, Technical and Medical business to form Wiley-Blackwell. Registered offi ce: John Wiley & Sons, Ltd, The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK Editorial offi ces: 9600 Garsington Road, Oxford, OX4 2DQ, UK The Atrium, Southern Gate, Chichester, West Sussex, PO19 8SQ, UK 111 River Street, Hoboken, NJ 07030-5774, USA For details of our global editorial offi ces, for customer services and for information about how to apply for permission to reuse the copyright material in this book please see our website at www.wiley.com/wiley-blackwell The right of the author to be identifi ed as the author of this work has been asserted in accordance with the UK Copyright, Designs and Patents Act 1988. All rights reserved. No part of this publication may be reproduced, stored in a retrieval system, or transmitted, in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, except as permitted by the UK Copyright, Designs and Patents Act 1988, without the prior permission of the publisher. -

Distance of Antenna from Neighboring Houses (Feet): /UO' Distance of Antenna from Power Distribution Line Or Equipmmt
I' J Distance of antenna from neighboring houses (feet): /UO' Distance of antenna from power distribution line or equipmmt I I Made "---"- ------ .. ..*- . I I.. c .r qe * Page 1 of 2 +* - es Burtle ,: ,: Thursday, September 02,2004 258 PM James Burtle; Alan Scrim; Alan Stillwell; Bruce Franca; Karen Rackley iect Fw: BPL Interference complaint, new Ion-public: Fa M US^ OI@ *** Mginal Mesage- n: Doyle Hady [mailbo:dghardy@chw*net] i: Thursday, Septiember 02,2004 236 PM 4nh Wridt je.. BPL 1- le of complainant: Doyle.Hardy sign (if applicable): KSHTE ion location: 6521 Circleview Dr ,Ft Worth Tx, 76180 ling address (ifdifferen): 1, state, zip: Ft Worth TX, 76180 =phone: 817 -281-8670 Fm-d:[email protected] miption of Interference:- I was in contact with -K8NDS who at the time was opemting mobil station :ottonwood AZ. He was at Murphy’@ in Cottonwood. The interfence i hmthe local BPL jest made my signal unreadable. When the BPL was off I had a signal of S7 and very scription of station: My station is Kenwood TS-570D, running 100 ‘g of inwcrence: Dak Time Frequency Receive Interfering Description Mode signal 09/02/04 18OOGMT 14225 SSB 57 + was informedthat MHZ BPL totally made my signal unreadable. I I - :sage page 2 of 2 *' 8 I * es protect from harmful interference 's rules, Part 15 includes a definition of harmful interference. It can be found in S15.3 : "Harmful interference. Any emission, radiation or induction that endanger8 the functioning ). a radio navigation service or of other safety services or seriously degrades, obstructs, or eatedly interrupts a radiocommunication service operating in accordance with this chapter." < rules are very clear about the operation of Part 15 devices, too. -

TNM Classification of Malignant Tumours
Table of Contents Cover Title Page Preface References Acknowledgments Organizations Associated with the TNM System Members of UICC Committees Associated with the TNM System Section Editors Introduction References Head and Neck Tumours Lip and Oral Cavity Pharynx References Larynx Nasal Cavity and Paranasal Sinuses Unknown Primary – Cervical Nodes Malignant Melanoma of Upper Aerodigestive Tract Major Salivary Glands Thyroid Gland Digestive System Tumours Oesophagus Stomach Reference Small Intestine Appendix Colon and Rectum Anal Canal and Perianal Skin Liver Intrahepatic Bile Ducts Gallbladder Perihilar Bile Ducts Distal Extrahepatic Bile Duct Ampulla of Vater Pancreas Well Differentiated Neuroendocrine Tumours of the Gastrointestinal Tract Lung, Pleural, and Thymic Tumours Lung References Pleural Mesothelioma Thymic Tumours References Tumours of Bone and Soft Tissues Bone Soft Tissues Gastrointestinal Stromal Tumour (GIST) Skin Tumours Carcinoma of Skin (excluding eyelid, head and neck, perianal, vulva, and penis) Skin Carcinoma of the Head and Neck Carcinoma of Skin of the Eyelid Malignant Melanoma of Skin Merkel Cell Carcinoma of Skin Breast Tumours Reference Gynaecological Tumours Reference Vulva Vagina Cervix Uteri Uterus – Endometrium Reference Uterine Sarcomas References Ovarian, Fallopian Tube, and Primary Peritoneal Carcinoma Reference Gestational Trophoblastic Neoplasms Reference Urological Tumours Penis Prostate References Testis Kidney Renal Pelvis and Ureter Urinary Bladder Urethra Adrenal Cortex (ICD O 3 C74.0) Ophthalmic -

Las Vegas Wash Time-Of-Travel Study
Las Vegas Wash Time-of-Travel Study Research and Development Office Science and Technology Program Final Report ST-2016-2262-1 U.S. Department of the Interior Bureau of Reclamation Research and Development Office June 2016 Mission Statements The U.S. Department of the Interior protects America’s natural resources and heritage, honors our cultures and tribal communities, and supplies the energy to power our future. The mission of the Bureau of Reclamation is to manage, develop, and protect water and related resources in an environmentally and economically sound manner in the interest of the American public. REPORT DOCUMENTATION PAGE Form Approved OMB No. 0704-0188 T1. REPORT DATE May 2016 T2. REPORT TYPE Research T3. DATES COVERED 10-01-2015 to 6-30-2016 T4. TITLE AND SUBTITLE Las Vegas Wash Time-of-Travel Study 5a. CONTRACT NUMBER RY1541EN201522262 5b. GRANT NUMBER 5c. PROGRAM ELEMENT NUMBER 1541 (S&T) 6. AUTHOR(S) 5d. PROJECT NUMBER Blasius, Becky, Lower Colorado Region, Resources Management Office 2262 Kirsch, Janet, Lower Colorado Region, Resources Management Office (retired) 5e. TASK NUMBER Danner, Allison, Lower Colorado Region, Engineering Services Office 5f. WORK UNIT NUMBER LC-2628 7. PERFORMING ORGANIZATION NAME(S) AND ADDRESS(ES) 8. PERFORMING ORGANIZATION Bureau of Reclamation, Lower Colorado Region, Resources Management Office REPORT NUMBER Bureau of Reclamation, Lower Colorado Region, Engineering Services Office ESO Project Number 2016-003 PO Box 61470, Boulder City, NV 89006 9. SPONSORING / MONITORING AGENCY NAME(S) AND ADDRESS(ES) 10. SPONSOR/MONITOR’S Research and Development Office ACRONYM(S) U.S. Department of the Interior, Bureau of Reclamation, R&D: Research and Development Office PO Box 25007, Denver CO 80225-0007 BOR/USBR: Bureau of Reclamation DOI: Department of the Interior 11. -
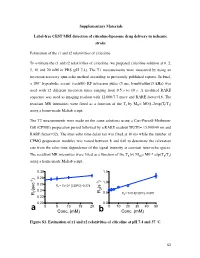
Conc. (Mm) B Conc
Supplementary Materials Label-free CEST MRI detection of citicoline-liposome drug delivery in ischemic stroke Estimation of the r1 and r2 relaxivities of citicoline To estimate the r1 and r2 relaxivities of citicoline, we prepared citicoline solution at 0, 2, 5, 10 and 20 mM in PBS (pH 7.4). The T1 measurements were measured by using an inversion recovery spin-echo method according to previously published reports. In brief, a 180° hyperbolic secant (sech80) RF inversion pulse (5 ms, bandwidth=15 kHz) was used with 12 different inversion times ranging from 0.5 s to 10 s. A modified RARE sequence was used as imaging readout with 12,000/7.7 msec and RARE factor=16. The resultant MR intensities were fitted as a function of the TI by MTI= M0(1-2exp(TI/T1) using a home-made Maltab script. The T2 measurements were made on the same solutions using a Carr-Purcell-Meiboom- Gill (CPMG) preparation period followed by a RARE readout TR/TE= 15,000/40 ms and RARE factor=32). The inter-echo time delay tau was fixed at 10 ms while the number of CPMG preparation modules was varied between 8 and 640 to determine the relaxation rate from the echo time dependence of the signal intensity at constant inter-echo spaces. The resultant MR intensities were fitted as a function of the TE by MTE= M0 * exp(TE/T2) using a home-made Maltab script. 0.30 1.5 ) 0.28 ) -1 1.0 0.26 -4 -1 R1= 7x10 [CDPC]+0.274 (s 2 (sec 1 0.24 R 0.5 R = 0.014[CDPC]+0.591 R 2 0.22 0.20 0.0 a 0 5 10 15 20 0 10 20 30 40 50 Conc.